AlphaFold 3 is the latest version of DeepMind’s AI model for predicting biomolecular structures. Unlike AlphaFold 2, it can model not just proteins, but also complexes with DNA, RNA, and small molecules. It represents a major leap forward in structural biology and drug discovery.

AlphaPulldown is a customized implementation of AlphaFold-Multimer designed for customizable high-throughput screening of protein-protein interactions. It extends AlphaFold’s capabilities by incorporating additional run options, such as customizable multimeric structural templates (TrueMultimer), MMseqs2 multiple sequence alignment (MSA) via ColabFold databases, protein fragment predictions, and the ability to incorporate mass spec data as an input using AlphaLink2.
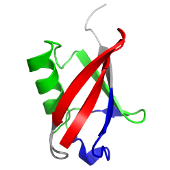
BindCraft is a computational tool designed to predict and model protein–ligand interactions. It integrates structural data and AI-driven methods to identify binding sites and estimate binding affinities. BindCraft is useful for drug discovery, molecular docking, and interaction analysis.
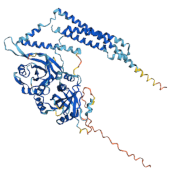
Boltz-2 is a next-generation deep learning model for predicting biomolecular structures and binding affinities. It surpasses AlphaFold3-level accuracy by jointly modeling complex interactions, including docking and affinity estimation. As the first model to rival physics-based FEP methods while being 1000x faster, Boltz-2 enables practical and accurate in silico drug screening.
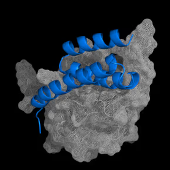
Chai-1 is a deep learning model for predicting RNA interaction sites at high resolution. It supports a wide range of partners, including proteins, DNA, and small molecules. Chai-1 enables detailed mapping of biomolecular interfaces for functional analysis and design.
ColabFold is an easy-to-use platform for predicting protein structures and complexes using AlphaFold2 and AlphaFold2-Multimer. It streamlines the workflow by automatically generating sequence alignments and templates with MMseqs2 and HHsearch.

ProteinMPNN is a deep learning model for protein sequence design based on fixed backbone structures. It generates amino acid sequences that are likely to fold into a given 3D structure. ProteinMPNN is widely used for protein engineering and de novo protein design.

RFdiffusion is a deep generative model for protein design based on diffusion probabilistic modeling. It builds 3D protein structures from scratch or around functional motifs with high flexibility and control. RFdiffusion is a powerful tool for creating novel proteins with desired shapes and functions.
HHalign-Kbest is useful to automatically obtain optimized alignments and models in case of low sequence identity (<35%) between a query and a template protein. It can generate k suboptimal (e.g. top-k scoring) alignments rather than only the optimal one which may contain small to large errors.
HHalign.KBest: exploring sub-optimal alignments for remote homology comparative modeling
submitted.

iSuperpose performs the 3D superposition of protein structures by best superimposing the alpha-carbons (or the backbone) of the proteins given a alignment specifying the correspondence between the structures. If no alignment is provided, a structural alignment will be calculated using TMalign. One the alignement is identified, the superposition is achieved using a quaternion based procedure using a specific eigen value calculation implementation. See QBestFit.
XmMol is a desktop macromolecular visualization and modeling tool designed to be easy to use, configure and enhance. Its graphics are based on X11, and part of its user interface is based on Motif. Thus it provides a way of displaying structures on any X11 server. Its main features are:
XmMol: an X11 and motif program for macromolecular visualization and modeling.
J Mol Graph. 1995 Feb;13(1):67-72, 62.
| af_analysis | Python package for the analysis of AlphaFold protein structure predictions. | View on Github |
| docking_py | Python library allowing a simplified use of the Smina, vina, qvina2 and qvinaw docking software. | View on Github |
| Fasta | Python library to manage single and multi fasta sequences (Python 2 version). | View on Gitlab |
| Fasta3 | Python library to manage single and multi fasta sequences (Python 3 version). | View on Gitlab |
| Gromacs_py | Python library allowing a simplified use of the Gromacs MD simulation software. | View on Github |
| Mol2 | Python library to manage simple and multiple mol2 files. | View on Gitlab |
| PyPDB | A python parser for PDB files (Python 2 version). | View on Gitlab |
| PyPDB3 | A python parser for PDB files (Python 3 version). | View on Gitlab |
| sdf | A python parser for sdf files. | View on Gitlab |

| BCscore |
A score based on Binet-Cauchy kernel. Allows for the search of both similar and mirror conformations. Addresses two major issue of the widely used root mean square deviation (RMSD):
|
Download |
| QBestFit | 3D superposition module, in C. Uses the quaternions. Very reliable. | Download |

TEF is an open-source software for decomposing protein structures into simpler yet informative units named Tightened End Fragments (or closed loops), which can be studied independently to understand protein architecture, folding, and evolution.
Distribution of tightened end fragments of globular proteins statistically matches that of topohydrophobic positions: towards an efficient punctuation of protein folding?
Cell Mol Life Sci. 2001 Mar;58(3):492-8.