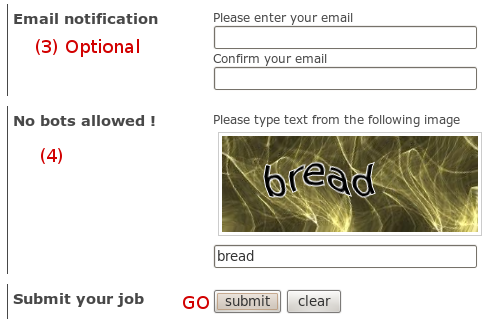
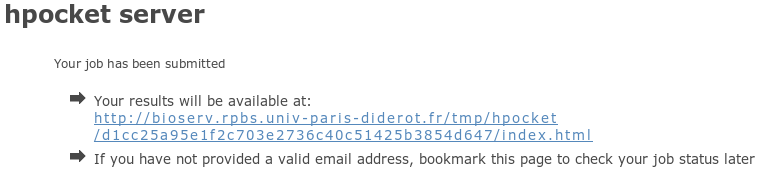
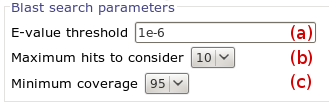
We use the same output page to display results of hpocket
job for both server. Before describing it, we just describe
for both servers, how to reach this page. Don't worry it's
really easy.
Getting to the results page
Default server
Just follow the
input tutorial,
where we describe all intermediates pages from which you
will be able to reach the results final page.
Advanced server (Mobyle)
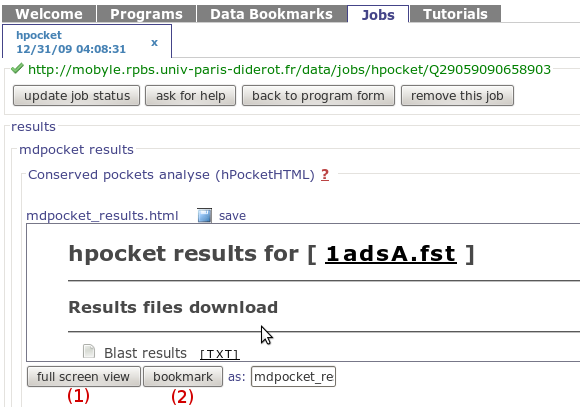
In mobyle, once your job will be finished, you will be
redirected to the mobyle results page. This results page
is shown in the snapshot on the right. The results page
is actually contained in the "Conserved pockets analyse
alpha spheres (hpocket HTML)" labeled scroll pane. To
have a more friendly view and reach the output pages
described just click on the Full screen view
button (1)
You also have the possibility to bookmark the result page
using the Bookmark button
(2) (enter the bookmark
name in the field on left)
Note that main program output results are provided both
in the final results page and in the mobyle interface.
This redundancy is comfortable to (i) analyse your results
in a specific and integrated web page and (ii) to directly
use the mobyle pipelining feature for further analysis.
<Back to top>
Results page description
The results can be roughly divided in 3 sections:
output files, snapshots and Visualisation.
Output files

Several hpocket output files are provided and described
bellow. You can download all of them.
-
Blast results
(1):
a text report for blast search.
-
PDB hits sequence aligment
(2):
a fasta file containing aligned homologous sequences
-
Superposed selected PDB hits archive
(3):
an archive containing all superposed homologous
structures (PDB).
-
mdpocket grid
(4):
it is the mdpocket output grid that stores density
information for each grid point. To learn more
about hpocket methodology and output, go tho the
hpocket method section
in the homepage, or read
the documentation.
-
Pockets grid point
(5): This file
contains all grid points having 3 or more Voronoi
Vertices in the 8A3 volume around the grid point
for each homologous hit. This output should be used
to define a specific zone (a pocket) on which you
may want to make further analysis using mdpocket
(not available in the hpocket server though, you
will have to run it yourself using the
desktop package).
-
Pocket density
(6): This file
contains the query protein, written as PDB file,
with the B-Factor values representing alpha spheres density.
Such a file allows intuitive, coloured visualisation
similar to that of the snapshots described below.
<Back to top>
Snapshots
Two set of snapshots are provided here: the first
set (left picture) represents the superposition of
all retrieved homologous structures, and the
second set (right picture) represents the query
structure surface coloured by alpha spheres density.
Here, coulours range fromblue (low density = no
particular cavity at this place) to red (high
density = hot spot = conserved cavity!).

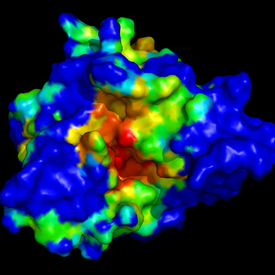
Note that you may obtain such display by dowloading
output PDB file "Pocket density" described previously,
and display it using pymol and VMD (color the molecular
surface by Bfactor).
<Back to top>
Visualisation
Currently, the visualisation is made using both Jmol and
OpenAstex, in which the mdpocket output PDB file is
automatically loaded in each viewer
(1) (query protein).
Using Jmol, you can view the Grid file
extracted from mdpocket results. A slider is provided to
change isovalue: the highest the isovalue is, the more
conserved is the corresponding cavity.
Using OpenAstex, the visualisation is
atom-centered. That is, the isovalues have been mapped
from the grid to the atoms and transformed to be somehow
B-factor-like for coloring purposes. The highest the
B-factor is, the more conserved is the cavity associated
with atoms.
Both visualisation methods use different metrics.
Isovalues will tipically range from 0 to N, with N having no real
limitation (depends on the number of snapshots; the slider
is limited to 800), while B-factor will range from 0 to 7-8,
as it is log-scaled. We are investingating a way to get
a common metric, and to merge these visualisation features
in a single viewer.
Jmol
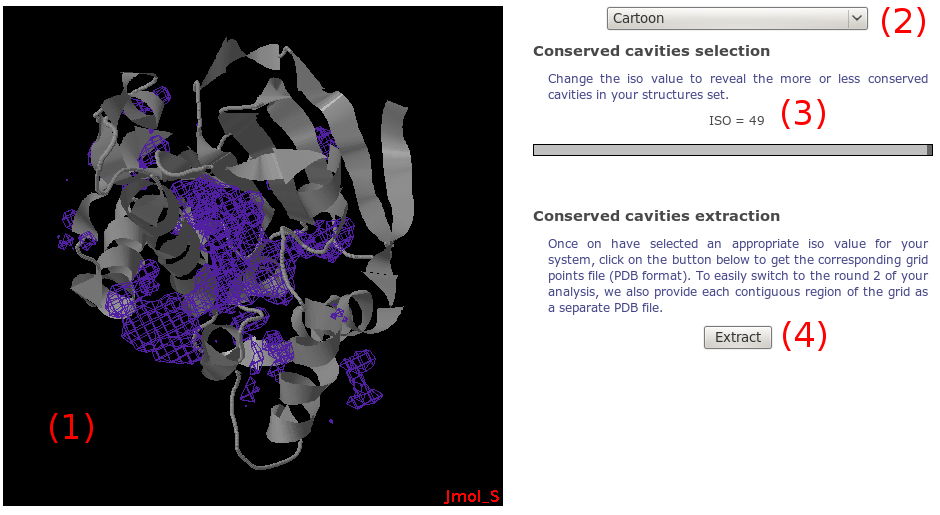
Using Jmol, the density grid is loaded along with the protein
(1).
On the right, you have a set of graphic components to
facilitate the viewing. A simple selection box
(2) allows you to perform
basic changes to the whole system representation
(display protein as cartoon, reset view...).
Using the slider on the right, you can change
(3) the so called "isovalue".
For a given grid point, this isovalue represents the number
of alpha spheres seen for all snapshots within a 8A radius.
Thus, a high isovalue will display protein cavities which are
highly conserved among homologous structures, while low
isovalues will rather show these conserved zones plus
other less conserved zones, which might be pockets specific
to one or several proteins.
As for mdpocket,
we give you the opportunity to downlad grid points
(PDB format) corresponding to each contigous points that
could form a potential pocket(4).
As there is no point to run the second round of mdpocket
to homologous structures, you may use these files only
to check how and where the conserved or transiant zones
are located on each homologous structures.
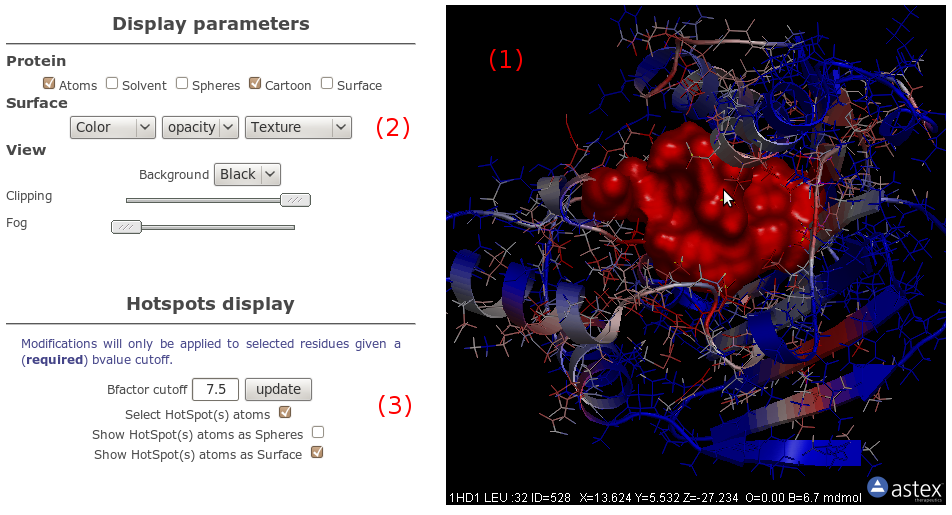
OpenAstexViewer
Using OpenAstex, atoms are coloured by B-factor, with the same
convention as that used for snapshots, that is, the more an
atom is close to red (resp. blue), the more conserved is
its corresponding cavity. Only here, the grid isovalue have
been mapped on atoms (see accompagning paper for formula)
so you can see directely which atom is associated with a
conserved zone.
In other words, red atoms may be part of common cavities
among homologous, while blue atoms may be part of specific
(or inexistant) cavities.
Besides basic visualisation options (2),
we give you the possibility to select atoms based on B-factor
value (3). By checking the
appropriate checkbox, all atoms having B-factor up to the
specifiede value (in the text field) will be selected.
If you change this isovalue cutoff, you have to click the
update button to update atom selection.
0 is the minimum B-factor value, while the maximum should
lie around 7 or 8 (due to log-scaling of isovalues).
Remember that if you know Jmol and OpenAstex, you can
access the display popup menu by right clicking on the view.