Welcome to the SA-Frag service!
SA-Frag is a service that will, given an amino acid sequence, return 3D fragments predicted to match the various positions of the sequence. SA-Frag will thus return an alignment of the fragments identified with the query and a collection of 3D structures corresponding to the fragments in the PDB format.
SA-Frag relies on a structural alphabet profile comparison approach. The input sequence will be converted to a SA profile used to mine large collections of proteins. The output will consist in PDB fragments having similar profiles and their position in the query. A systematic search is performed for fragments of size from 6 to 27 amino acids, over all positions of the input sequence. Several clustering and redundancy elimination filters allow the identification of a limited number of fragments.
 |
 |
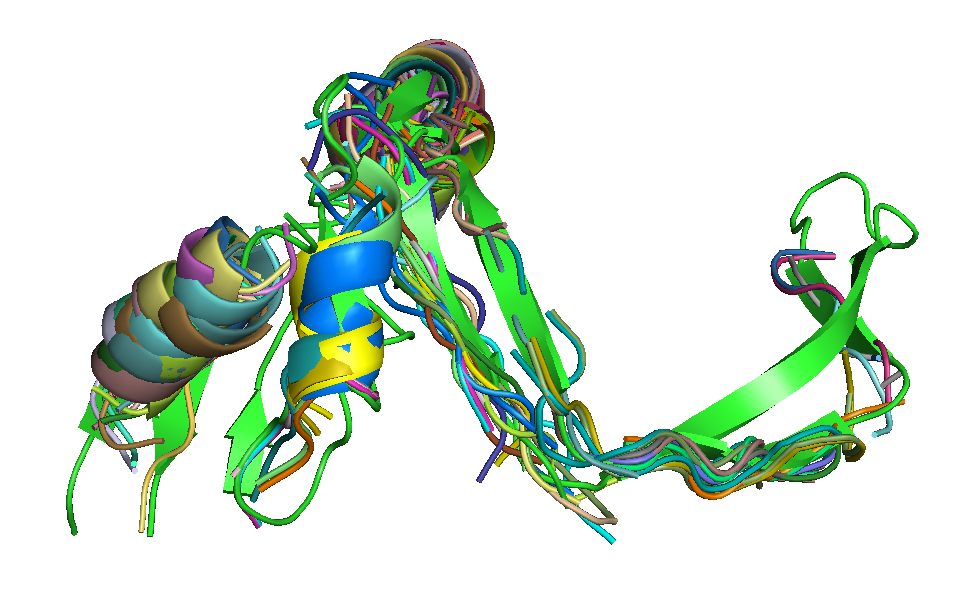 |
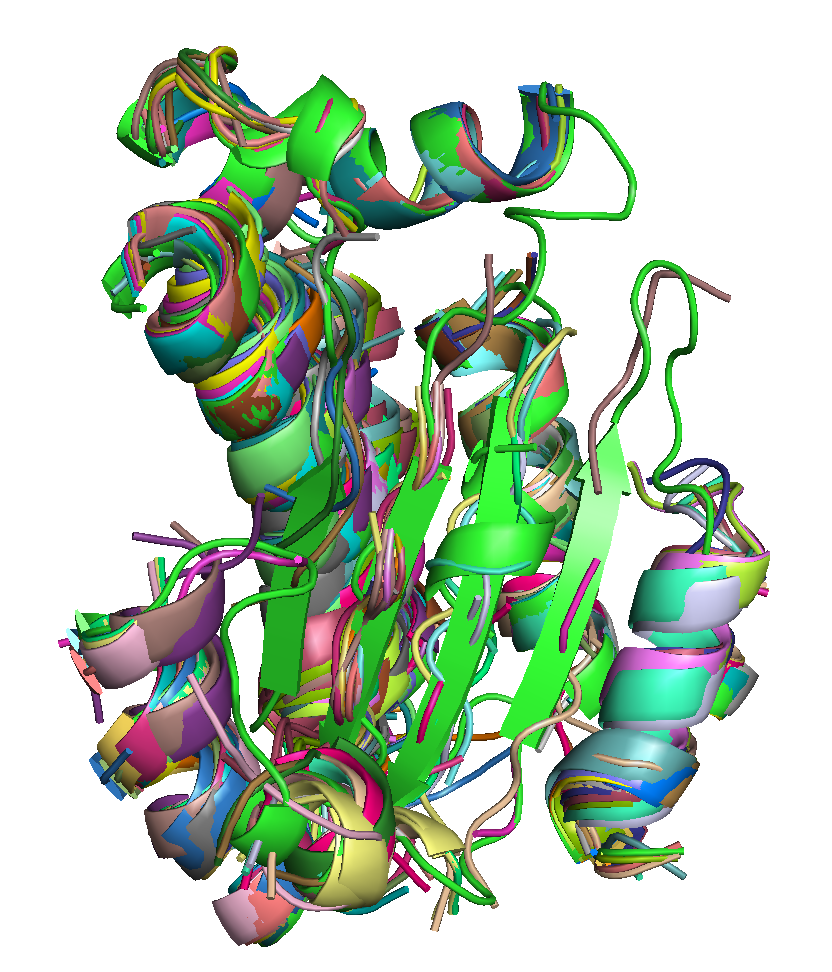
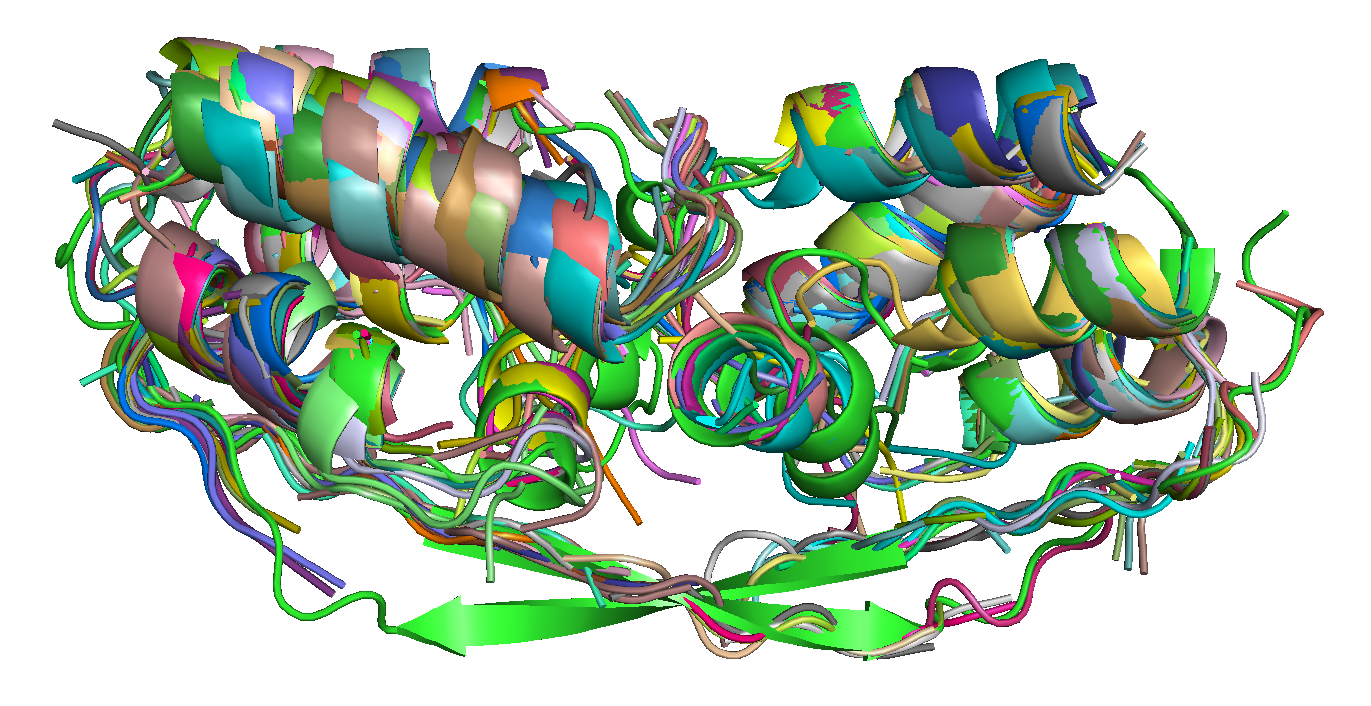
| T0684 FM domain. | T0734 FM domain. | T0741 FM domain. |
Access the
SA-Frag server @ the
RPBS Mobyle Portal.
When using this service, please cite the following reference:
Detecting protein candidate fragments using a structural alphabet profile comparison approach.
PLoS One. 2013 Nov 26;8(11)
When using this service, please cite the following reference:
Detecting protein candidate fragments using a structural alphabet profile comparison approach.
PLoS One. 2013 Nov 26;8(11)
Features
- Reference structure: SA-Frag accepts a reference PDB file to return fragments superimposed on it. (see usage).
- Processing time: Execution time is a function of the size of the sequence. Once the processing has started, it can vary from some minutes to more than one hour, depending on the server load.
Limitations
- Amino acid sequence size: In theory, there is no limit for the sequence size, but huge sequences have a prohibitive cost. Thus the server will not consider sequences of more than 1000 residues.
- Fragment identification: Fragment search is assumed using a gapless approach.
A step by step tutorial
-
Input the query sequence
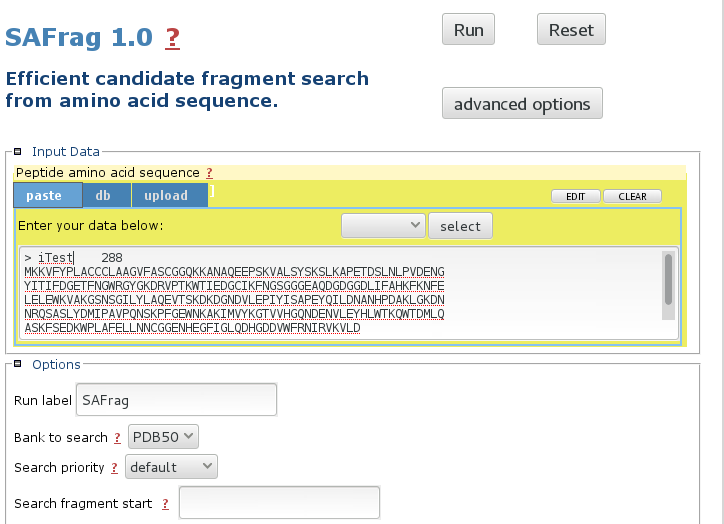
The mobyle interface allows to paste, upload the sequence. The default bank to search is the PDB50 subset. More details about the options in the Usage section. -
Launch the calculations
Click run to launch the calculations.
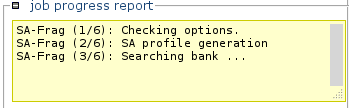
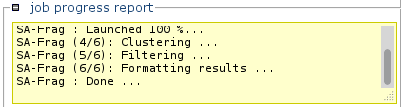
Information about the status of the processing will progressively appear.
Note: Calculations can be long. -
Browse the results
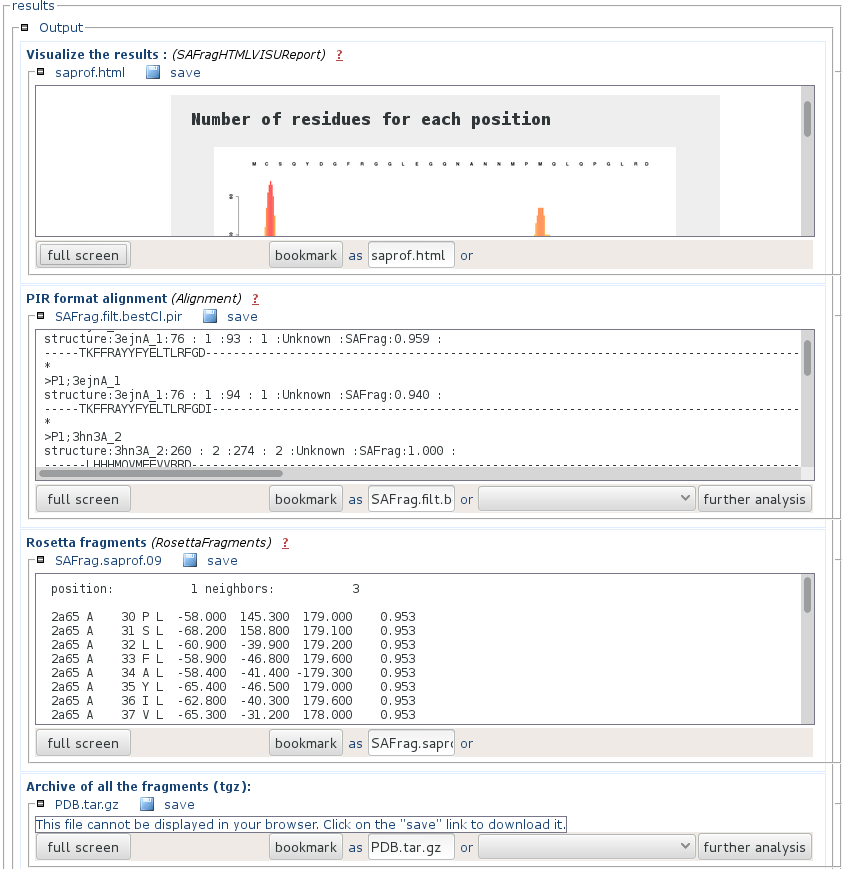
- "Visualize the results" is an html page that presents a synthetic view of the number of results at each position of the query, and the alignement of the query and the identified fragments (results). You can get a full page visualization by clicking the "full screen" button.
- "Pir formatted alignment" is the alignment file ready to use for Modeller. You can download it by pressing the "save" button.
- "Rosetta fragments" is a presentation of the results using the Rosetta format.
- "Archive of the fragments" contains the PDB fragments of the identified fragments. File names match that of the PIR formatted alignment. You can download it by pressing the "save" button.
Usage
Input
- Input sequence: Input sequence file must be in FASTA format. The query peptide sequence must contain a string of only the 20 standard amino acids in uppercase, using the 1 letter code. Sequence length is presently limited to 1000 amino acids
Input options
- Run label: A title for your run. It MUST be a single word (no spaces, no special characters). It will be used to generate the name of the models.
- Bank to search: The proposed banks correspond to a collection of 4649 proteins at less than 25% sequence identity, associated with the HHfrag publication (PDB25), non redundant collections of proteins at 30 and 50% sequence identity (PDB30 and PDB50).
- Search priority: To some extent, it is possible to tune the returned fragments by favoring coverage("max. Cov.") or precision ("max. Prec.") compared to the default values. Increased coverage will result in decreased precision and conversely.
- Restraining the search: It is possible to narrow the search to a subsequence by specifying start and stop positions in the sequence. Fragments subjected to 3D modelling. This facility is useful for peptides where the N- and C- terminal regions are known to be disordered and the user wishes to focus on the modelling of the structured core. To this end, our tests show that the prediction of the SA profile is best using the complete sequence rather than a truncated sequence. The region that will undergo the 3D modelling must be specified on the form: x-y (e.g. 15-50), indicating that the 3D models will encompass the region between residues 15 and 50 (included), where 1 is the first residue of the input sequence.
- Reference structure: Input reference structure file must be in PDB format. This file must only contains a single model, with no hetero-residues and have the exact same length (in amino acids) as the query sequence.
Results
- PEP-FOLD produces informative report to guide the user through the best model selection.
-
Progress report
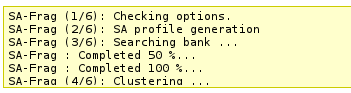 This section will incrementally provide information about job progression and errors if any.
A typical run should produce a report similar to that. Errors related to the input data specified are now also reported in this section.
This section will incrementally provide information about job progression and errors if any.
A typical run should produce a report similar to that. Errors related to the input data specified are now also reported in this section.
-
3D display
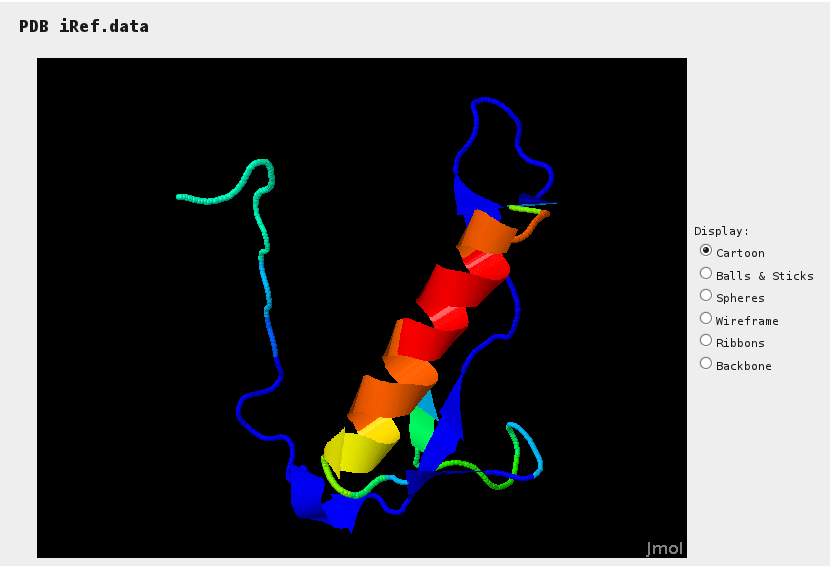 Jmol provides a mean to assess sections of the reference structure (if specified) for which hits have been identified. Colors range from blue (no hit) to red (many hits).
Jmol provides a mean to assess sections of the reference structure (if specified) for which hits have been identified. Colors range from blue (no hit) to red (many hits).
-
Hit effectives along sequence
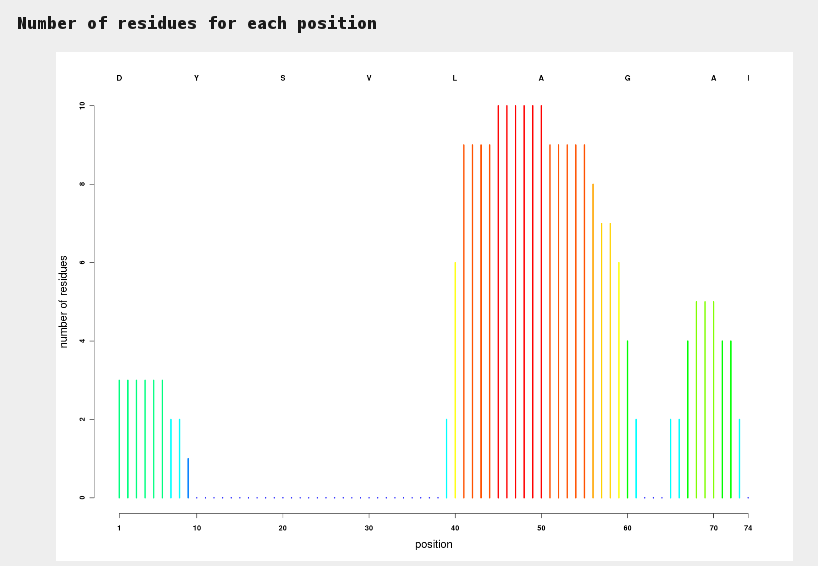 This plot allows to identify at a glance sections of sequences for which hits have been identified. Colors range from blue (no hit) to red (many hits).
This plot allows to identify at a glance sections of sequences for which hits have been identified. Colors range from blue (no hit) to red (many hits).
-
Aligned hits
 This area depicts the alignment of the fragments onto the query sequence.
Upper case letters stand for sequence identity with the query.
This area depicts the alignment of the fragments onto the query sequence.
Upper case letters stand for sequence identity with the query.
-
PIR results
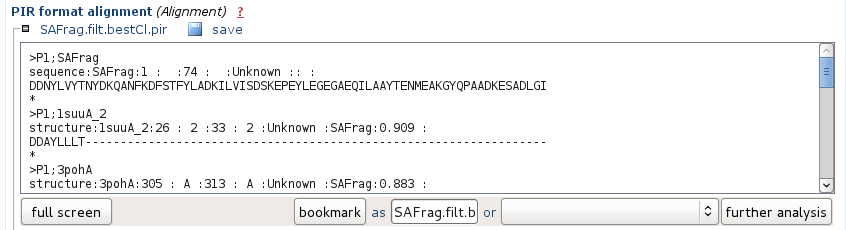 It corresponds to the alignment of the hits in the PIR format. The names correspond to the PDB archive, so as to be ready for use in a program such as Modeller for instance.
It corresponds to the alignment of the hits in the PIR format. The names correspond to the PDB archive, so as to be ready for use in a program such as Modeller for instance.
-
Rosetta fragment
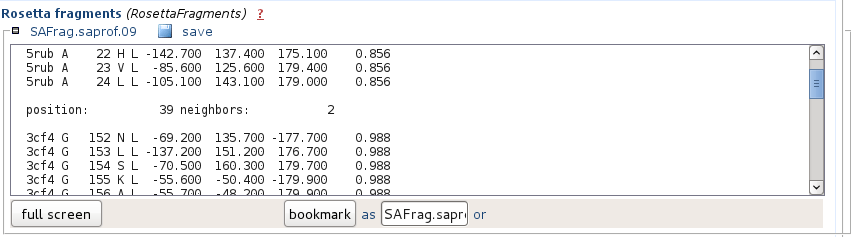 It corresponds to the hits in the Rosetta fragment format.
It corresponds to the hits in the Rosetta fragment format.
-
Fragment PDB archiveThis archive contains the PDB of the hits identified. It is in the unix tar format compressed using gzip.
Once saved on your computer, enter for instance (unix) tar xzf AllModels.tgz to inflate the archive.
Concepts
- Structural alphabet PEP-FOLD is based on the concept of structural alphabet [1] , i.e. an ensemble of elementary prototype conformations able to describe the whole diversity of protein structures.
History
- 2013, February Service 1.0 release
- 2013, January Service open for final tests. Minor adjustments in SA-Frag core parameters.
- 2012, September Implementation of several collections of structures (PDB30, PDB50)
- 2012, July First beta-version of the service
References
[1]
Detecting protein candidate fragments using a structural alphabet profile comparison approach.
PLoS One. 2013 Nov 26;8(11)
<Back to top>
Detecting protein candidate fragments using a structural alphabet profile comparison approach.
PLoS One. 2013 Nov 26;8(11)