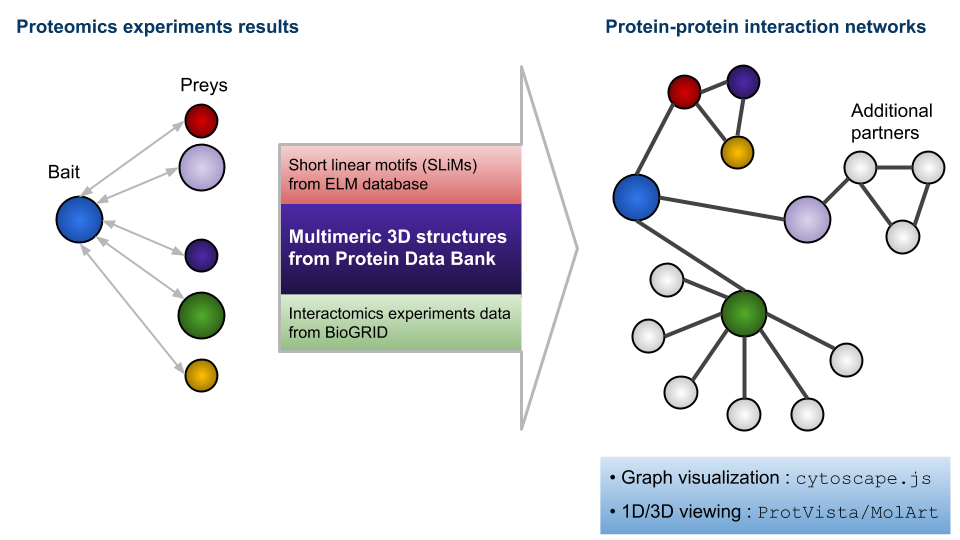
Template structures for each input sequence found by remote homology detection using HHsearch [2], followed by thorough HHsearch cluster expansion/analysis;
Interactive display/exploration of the inferred graph of interactions, through cytoscape.js [7].
Interactive 3D display of the template complexes, through MolArt [8].
Usage
Input
The main input for Proteo3Dnet is simply a list of candidate protein partners.
There is no need for this list to result from proteomics experiments. It can be any theoretical list of proteins for which users want to perform an interaction network analysis.
a series of valid protein sequences in the FASTA format.
For UniProt identifiers, AC and ID cannot be mixed. Therefore, it should either:
entry names
P73_HUMAN
P63_HUMAN
or accession numbers
O15350
Q9H3D4
For FASTA sequences, the UniProt identifier must be specified specified in each first FASTA line. This can be done in three different ways, which cannot be mixed:
WARNING: The number of submitted proteins cannot be greater than 400.
Other inputs of Proteo3Dnet are:
Mode: In the Normal mode, the homology search is performed using HHsearch. However, as many protein models are available in the SWISS-MODEL Repository, selecting the Fast mode will trigger instead the identification of pre-computed homologies from the SMR, which is must faster.
NOTE: The Fast mode can prove useful to provide a quick look at the data. However, SWISS-MODEL usually contains only models at a rather high sequence identity and, thus, significantly enhanced results are expected from the Normal mode in which more remote homology links can be detected.
Label: It will be displayed at the bottom of the result page, for users to identify their different jobs.
Organism (Optional): By specifying the name of a species in this field, only the input data belonging to that species will be considered. Mixing inputs from different species is not recommended. Otherwise, only the data of the most represented species, or that belonging to the organism specified in the advanced options, will be processed.
Max. BioGRID nodes (Optional): Users can define the maximum number of nodes that will be added (i.e. in addition to the input nodes) to the computed graph, by the integration of BioGRID data (by default, n=15).
Max. 3D nodes (Optional): Similarly to the above option, users can define the maximum number of nodes that will be added by the integration of 3D structural data (by default, n=50).
Min. ANCHOR2 score (Optional): The transient interactions analysis is based on both ELM motifs and detection of intrinsically disordered regions (IDRs) with IUPred2A. The higher its ANCHOR2 score, the more reliable the IDR predictions (which means fewer ELM edges in the graph).
Fold change (Optional): The value of the fold change (number of protein copies detected in the sample) for each candidate protein. It will be used to modulate the size of the nodes in the graph display of the results.
For fold changes in the [0, 1[ interval, the input nodes will be represented in red and their size will be proportional to the inverse of the fold change, i.e. nodes associated with FC values of 10 and 0.1 will have the same size, but the later will be colored in red. If null or negative values are provided for the fold change, Proteo3Dnet will assume that they correspond to the log(FC).
Usage: output
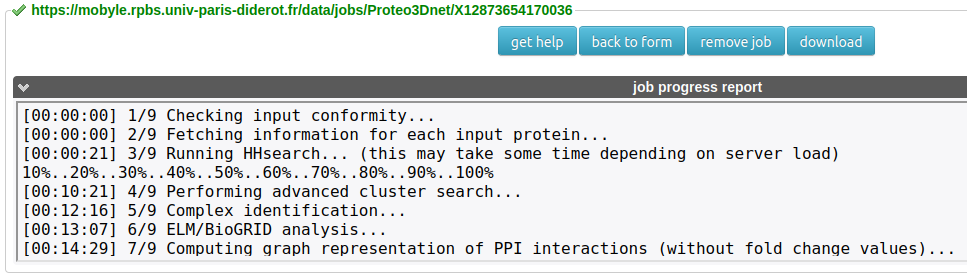
A. Progress report
This section will incrementally provide information about job progression and errors, if any.
A typical run should produce a progress report similar to the following:
Slight variations can occur depending on the version of the service.
Errors related to the input data specified are also reported in this field.
For a medium-sized dataset, such as our demo Pragmin interactome (N=62), the whole analysis will be achieved in ~15 minutes, in Normal mode.
Above the progress report is the URL of the job (in green), which can be used for accessing the results later.
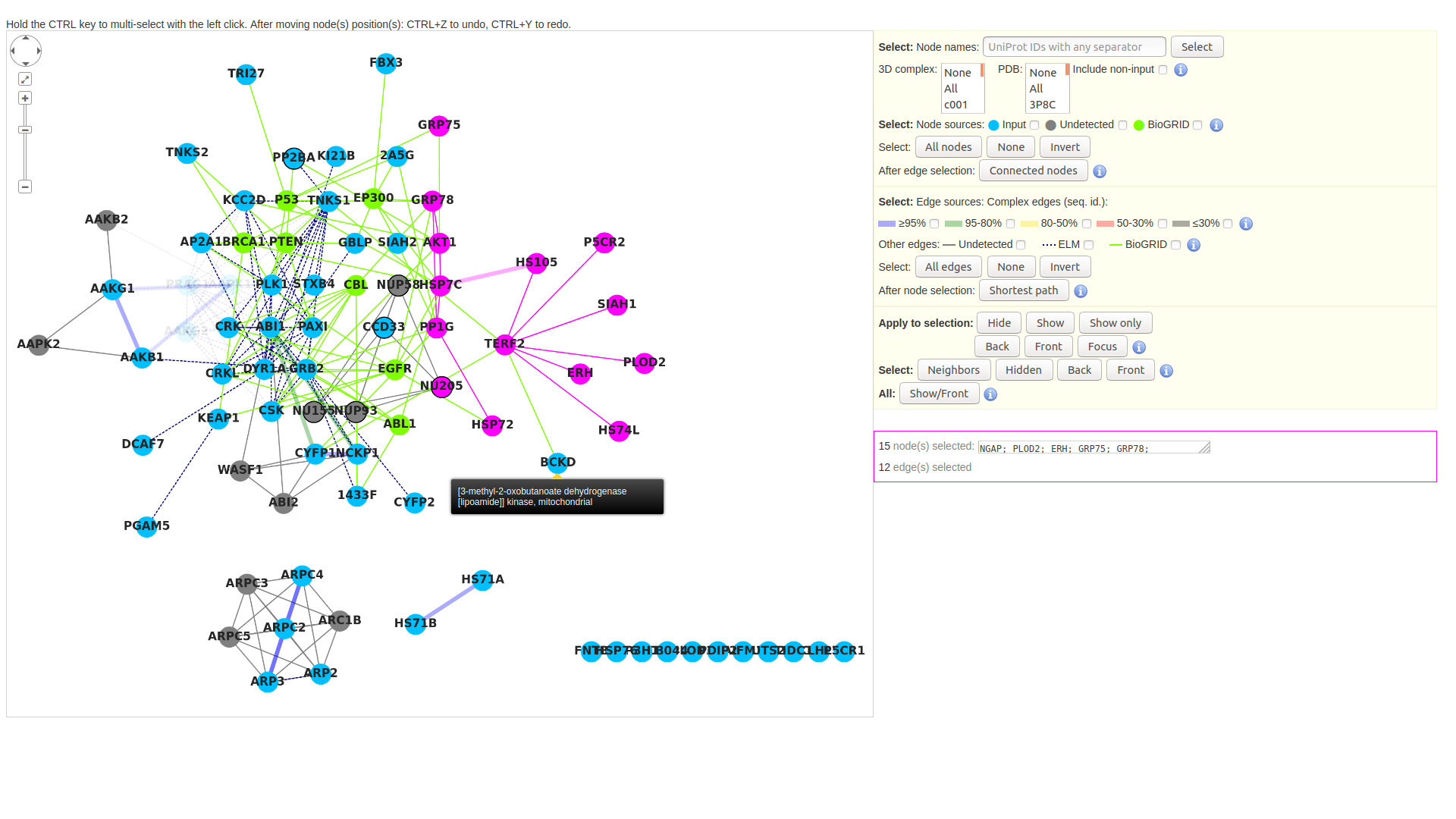
B. Protein-protein interactions: visualization
The results produced by the pipeline can be visualized as a graph, thanks to cytoscape.js. The nodes are of three types: input proteins, undetected partners, and BioGRID partners. The last result from the integration of the BioGRID data. Above the graph viewer are buttons which allow to select nodes and edges depending on their attributes.
IMPORTANT! Details on how to handle the graph are provided here: Tutorial
C. Links to external resources for graph nodes:
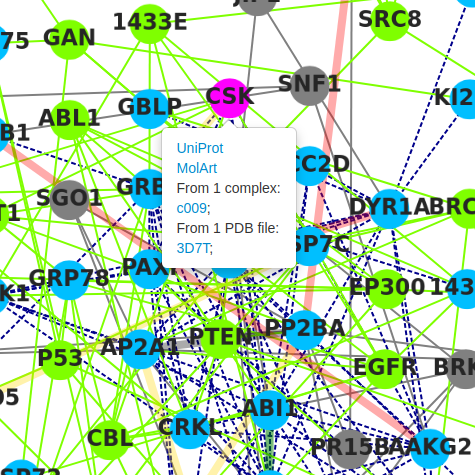
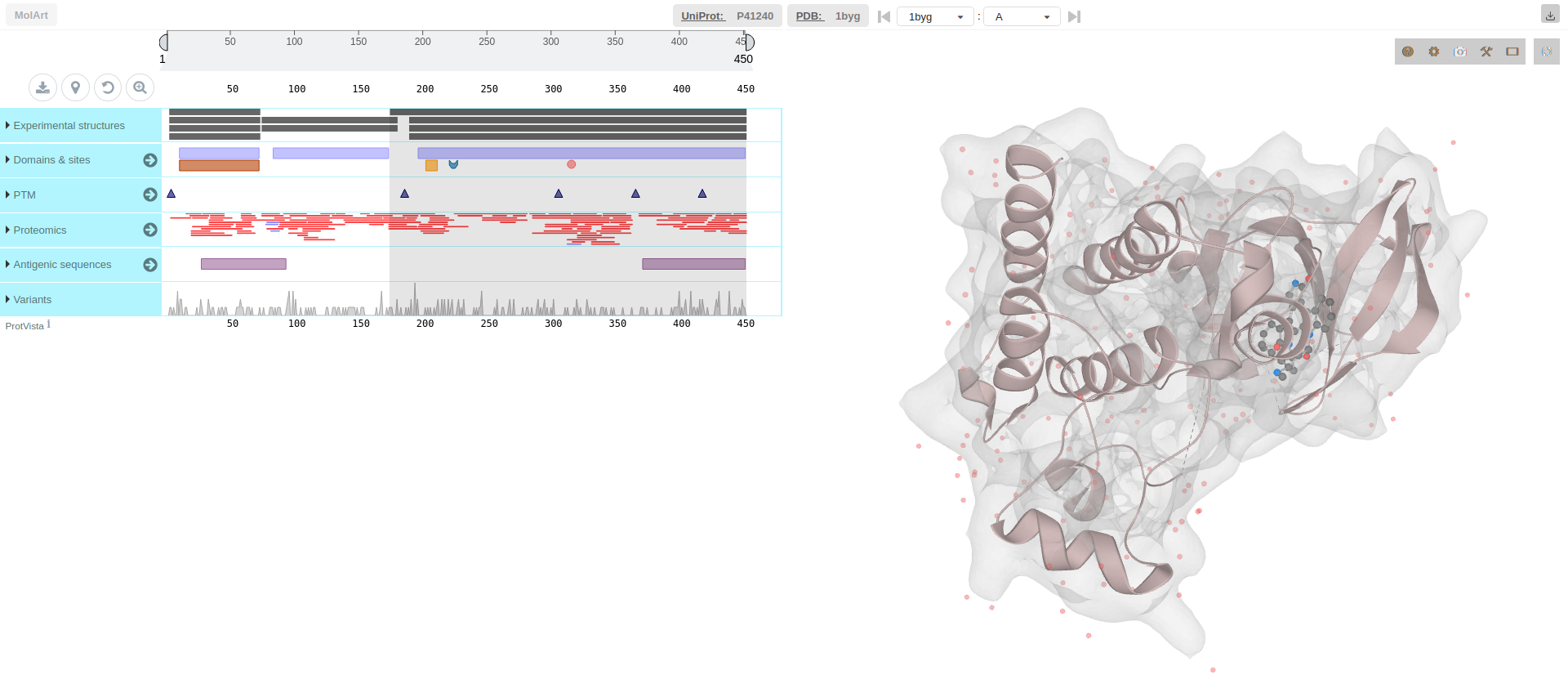
Right clicking on any node of the graph proposes to get further information, either opening the UniProt page related to the protein, or to visualize it using MolArt, which displays the 3D structure of the interaction partner. Thus, the protein chain can be visualized in the context of the oligomeric assembly. The MolArt viewer also provides various annotations of the protein sequence, depending on the data available.
For example, right clicking on the CSK node, in the result page of the preset data (Pragmin), and choosing MolArt, one gets redirected to the following page (for more information, please refer to the MolArt documentation).
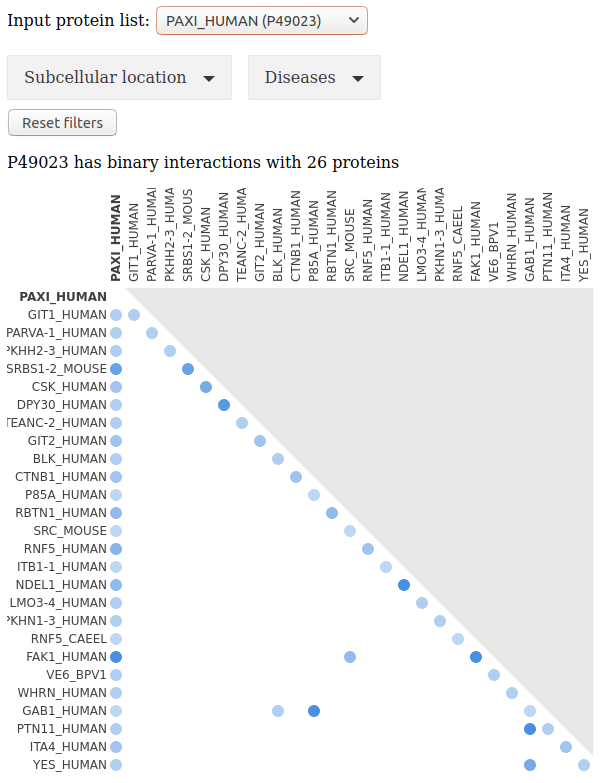
Select an entry (i.e. an input protein) to see the interactions described in IntAct.
Additional filters apply to subcellular localization and diseases.
The matrix summarizes the interactions between all experimentally verified interacting partners of the entry. Pairwise interactions described in the IntAct database are depicted using blue spots. Color intensity is a function of the number of experiments reporting the interaction. More information can be accessed by clicking on the spots.
D. Tables
For advanced users interested in structural modeling of multimeric proteins, details about the interolog detection and the subsequent analysis of 3D complexes are available in two tables. A third table about the detection of homo-oligomers, which is based on distant homologies, is also generated.
NOTE: More information about these tables are provided here.
Interpreting the results
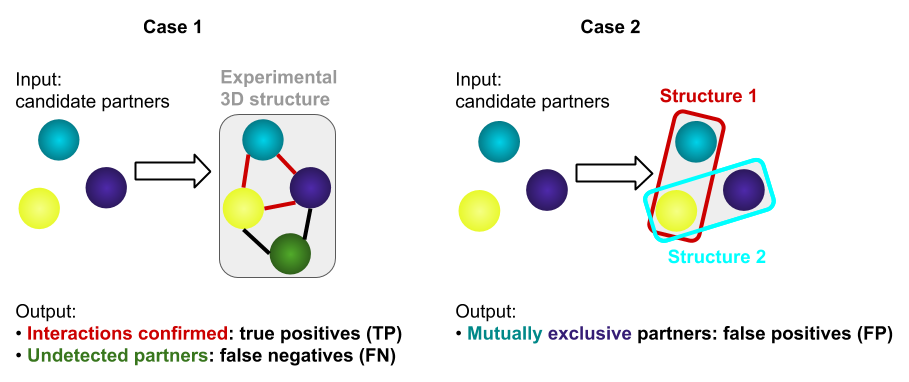
The data submitted by users may suffer from the presence of identified protein-protein interactions that do not occur physiologically (false positives, FP) and the non-detection of genuine associations (false negatives, FN).
After the search for 3D structures, each protein from the input list is associated to one, several, or no PDB entries. When several proteins from the submitted list share the same PDB entry (3D complex) (while corresponding to distinct protein chains), they are considered as true partners (true positives, TP). In this case, when the PDB entry found has one or several chains that are not present in the input list, these chains are then considered as potentially undetected partners (FN).
When two input proteins share a common partner while being seemingly mutually exclusive, this may help identifying FP: typically, subunits that are alternatively part of a complex, but do not interact with each other.
Thanks to the search for remote homologies, Proteo3Dnet will connect two input candidates, when the two corresponding homologous proteins are both found interacting within the same PDB structure. In such case, the interaction is not 100% confirmed, as the confirmation relies on the conservation of the interaction throughout evolution. That is why the sequence identity (%) is provided in the results produced by Proteo3Dnet (graph and tables).
Browser compatibility
OS
Version
Chrome
Firefox
Microsoft Edge
Safari
Linux
Ubuntu 20
87.0
84.0
n/a
n/a
MacOS
Mojave
87.0
84.0
n/a
12.0 ⚠
Windows
10
87.0
84.0
87.0
n/a
WARNING: For Safari 12.0 on MacOS Mojave, the graph visualization freezes when opening a MolArt window.
Project history
2020, Fall: User interface of the web server simplified.
2020, April: Method underlying Proteo3Dnet published in the Journal of Proteome Research (DOI: 10.1021/acs.jproteome.0c00066).
2019, December: Enhanced version of Proteo3Dnet with enhanced presentation of the ELM results.
2019, November: Enhanced version of Proteo3Dnet as a web server (binary interactions, more synthetic presentation of the results).
2019, November: MS2MODELS presentation at the SFCi meeting.
2019, November: MS2MODELS presentation at the MASIM workshop.
2019, October: Implementation of an interface to ELM.
2019, October: Many bug fixes related to the management of hhsearch clusters. Extensive test for several data sets on pragmin, proteasome and p73 MS interactome data.
2019, July: Proteo3Dnet as a web server in the RPBS mobyle portal. Embeds cytoscape.js and MolArt interfacing.
2019, April: MS2MODELS presentation at the GGMM congress.
2018, November: First implementation of full Proteo3Dnet pipeline.
2018, July: Project start.
2018, April: MS2MODELS project accepted.
2017, November: IFB call for pilot projects. Application of the MS2MODELS project to enrich MS interactome data in the light of available 3D structures.