PEP-FOLD 4
De novo peptide structure prediction considering pH and ionic strength variation.
De novo peptide structure prediction considering pH and ionic strength variation.
Pre-calculated results for(E)15, extremities blocked (see Batys et al., J. Phys. Chem. B, 2020 for experimental results).
• (E)15 at pH 7. Demo
• (E)15 at pH 3. Demo
When using this service, please cite the following references:
PEP-FOLD4: a pH-dependent force field for peptide structure prediction in aqueous solution.
Nucleic Acids Res., 2023, Web server issue.
A refined pH-dependent coarse-grained model for peptide structure prediction in aqueous solution.
Frontiers in Bioinformatics, 2023 3.
A Generalized Attraction-Repulsion Potential and Revisited Fragment Library Improves PEP-FOLD Peptide Structure Prediction.
J. Chem. Theory Comput. 2022 Apr 12;18(4):2720-2736.
PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex.
Nucleic Acids Res. 2016 Jul 8;44(W1):W449-54.
PEP-FOLD is a de novo approach aimed at predicting peptide structures from amino acid sequences. This method, based on structural alphabet SA letters to describe the conformations of four consecutive residues, couples the predicted series of SA letters to a greedy algorithm and a coarse-grained force field.
PEP-FOLD3 version available here . Based on sOPEP1, it uses a Hidden Markov Model sub-optimal conformation sampling approach faster by one order of magnitude than the previous greedy strategy, while not affecting performance. This makes possible the on-line generation of models for peptides from 5 to 50 amino acids in a few minutes. Consider this version to refine pre-existing models and/or to generate decoys keeping rigid regions of the structure, or to generate candidate conformations of peptide-protein complexes , by folding peptides on a user specified patch of a protein.
PEP-FOLD2 version available here is based on the greedy strategy can perform 3D modeling for linear peptides up to 36 amino acids, and allows user specified constraints such as disulfide bonds and inter-residue proximities.

PEP-FOLD4 relies on the sOPEP2 force field, a major evolution of sOPEP to model pH independent non bonded inteactions (see [6])
Presently, PEP-FOLD4 prediction is limited to amino acid sequences between 5 and 40 residues. For sizes more than 40 amino acids at neutral pH, users should consider using AlphaFold/ColabFold or related. For peptides shorter than 5 amino acids that are usually unstructured, conformational sampling approaches based on molecular dynamics simulations or approaches developped for small compounds should be preferred.
In date of december 2023, PEP-FOLD 3 only accepts the 20 usual amino acids. It will not process peptides with D-amino-acids, or unusual L- amino acids.
This field is to specify the amino acid sequence of the peptide. The input sequence file must be in FASTA format. The query peptide sequence must contain a string of only the 20 standard amino acids in uppercase, using the 1 letter code (see the pre-configured test example). The size of the input sequence can be as long as 50 amino acids.
A label for the files generated. It MUST be a single word (no spaces, no special characters). It will be used to generate the name of the models available for download.
Different suboptimal sampling algorithms can be used instead of the default forward backtrack algorithm (FBT) (see [4]). Forward-Backtrack should be considered for small sequences (up to 12 amino acids). For larger sequence, taboo-sampling (TS) should be considered, increasing the size of the taboo fragments for larger sequences. TS3 (resp. TS4, TTS5) stands for the non repetition of motifs of 3 (resp. 4, 5) consecutive SA letters (see [4]).
For short peptides, the generation of 100 models is generally enough to reach native or near native conformations. For larger peptides (more than 20 amino acids), generating 200 models is preferable.
Monte Carlo can be disabled by switching the number of steps to 0.
Former versions of PEP-FOLD used 700 K as best option. The sOPEP2 version makes it possible to lower to 370 K by default.
PEP-FOLD4 is fully reproducible for identical parameters. It is possible to generate different models by modifying the seed. Negative values redirect to the machine time.
This field is to reload the probabilities resulting from a previous run with the same sequence. If filled, these probabilities will be directly taken into account which will result in bypassing the local prediction step and save calculation time (around 40% time). Note: the input of probabilities not resulting from a previous run is strongly discouraged since it is most likely to result in spurious models.
PEP-FOLD4 is fully reproducible for identical parameters. It is possible to generate different models by modifying the seed. Negative values redirect to the machine time.
By default, the force field in use is that described in [6]. Switching Debye-Hueckel on will provide access to pH and ionic strenght parameters.
This is to specify the pH. It is only effective if the "Use Debye-Huckel" switch is turned to "Yes".
The ionic strength (mM). A default of 1 is in use by default. It is only effeective if the "Use Debye-Huckel" switch is turned to "Yes".
By default, the extremities are charged. It is possible to neutralize them by specifying which of the Nter (acetyl), Cter (N-methyl) or both extremities should be neutralized. This is only effective if the "Use Debye-Huckel" switch is turned to "Yes".
By default, the pKa values are assigned to standard values in aqueous solution, an approximation since pKa values may vary with the amino acid composition adn the conformation. It is possible to assign, for each residue in the sequence, a specific pKa value. One assignment is considered per line, in the form: residue position in the sequence (numbers from 1) - pKa value. For instance for a sequence like AAEVVKI, an input line of 3 5.6 would assign a pKa value of 5.6 to the glutamate at position 3 in the sequence. Lysine at position 6 would remain with its default pKa.
When selecting the load button, the sequence of the tau fragment will be loaded in the input sequence field and the Debye-Hueckel swith will be turned on.
PEP-FOLD main output consists in models. On-line interactive visualisation and model selection facilities are however proposed.

This section will incrementally provide information about job progression and errors if any. A typical run should produce a report similar to that. Errors related to the input data specified are now also reported in this section. For instance the report below explains a discrepancy between the sequence and the profile, probably due to an incorrect use of the "Probabilities from a previous run" field.

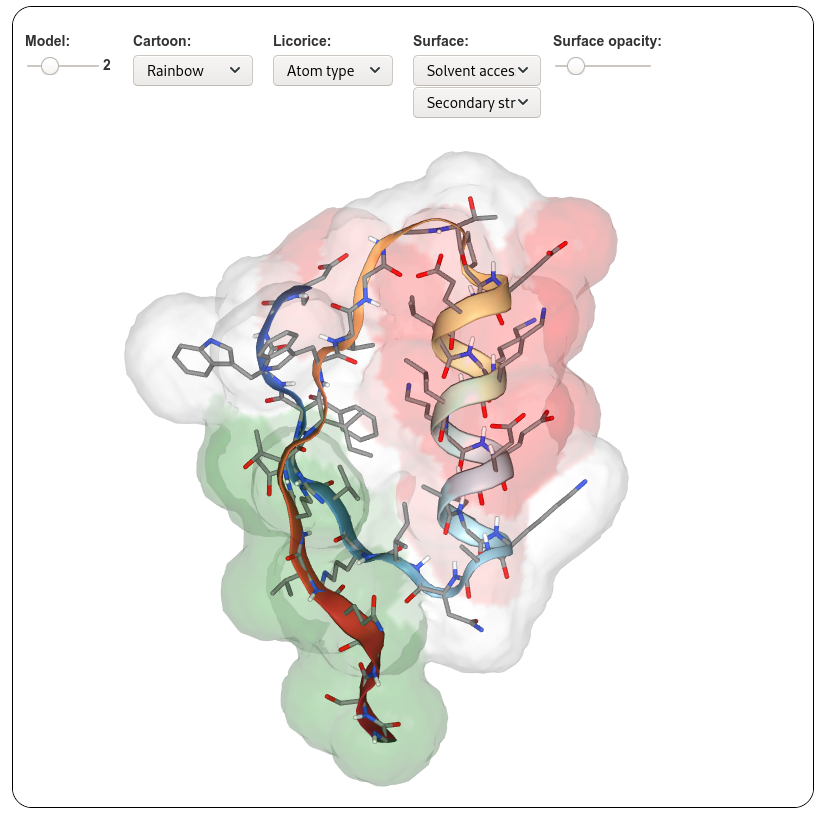
PEP-FOLD4 on-line interactive visualization of the models generated is based on the NGL javascript viewer . Different representations as well as colouring schemes can be selected. A menu makes possible to select a model among the 10 best models (representatives of the 10 best clusters).
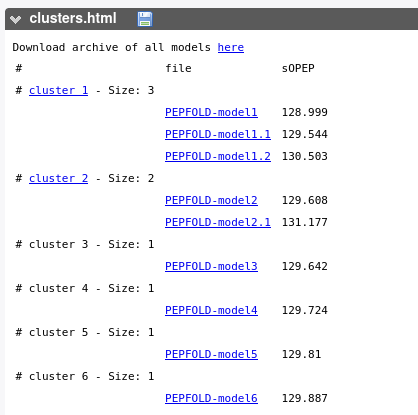
This report is primarily a table file that describes the clusters. For each model generated, the sOPEP energy of the model is reported.
Additionally, this table gives access to model download at different levels: The archive of all the models (top of table), archives of all models in a cluster - if the cluster contains several models, and each individual model.
The cluster ranks are defined according to their scores (sOPEP). The cluster representatives correspond to the models of the clusters having the best scores, i.e. with the lowest sOPEP energy. They are denoted as "modelx", where x is the rank of the cluster according to the sort key. When a cluster has several models, it is in turn sorted according to the sort key. The first model in the table is the representative, denoted on the form "modelx" and the following models are numbered using the "modelx.y" convention where x is the rank of the cluster and y the rank of the model in the cluster.
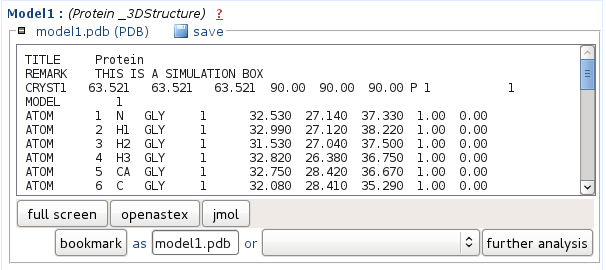
Representatives of the best 5 clusters predicted structure are provided in PDB format. You can either save the file onto your computer, or view it using NGL. In the Mobyle environment, PDB files can also be piped to other analyses such as the identification of secondary structures using stride or p-sea. For this, select the appropriate method beside the "further analysis" button, then launch it by clicking on "further analysis".
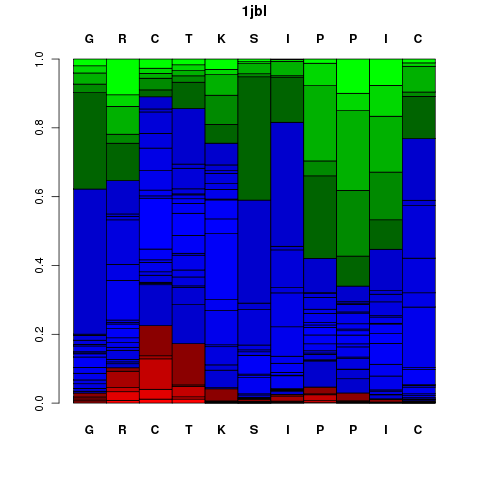
It corresponds to a graphical representation of the probabilities of each Structural Alphabet (SA) - see the Concepts - letter (vertical axis) at each position of the sequence (horizontal axis). Note that SA letters correspond to fragments of 4 residue length. The profile is presented using the following color code: red: helical, green: extended, blue: coil.
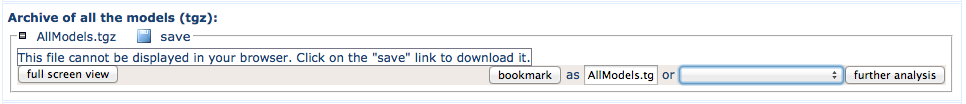
This archive contains all the models generated. It is in the unix tar format compressed using gzip.
Once saved on your computer, enter for instance (unix) tar xzf AllModels.tgz to inflate the archive.
As a simple test, you can either choose to:
Copy, paste the following sequence to the "Peptide amino acid sequence" field:
>1egs_A mol:protein length:9 GROES
TKSAGGIVL
or use Mobyle facilities :
Launch PEP-FOLD prediction, by clicking "Run" at the top of the page.
PEP-FOLD is based on the concept of structural alphabet [1], i.e. an ensemble of elementary prototype conformations able to describe the whole diversity of protein structures.
The sOPEP potential helps us to limit the roughness of the peptides energetic landscape, by simplifying side chains representation by a single bead. sOPEP2 parameters (free of Debye-Hueckel formalism) were optimized using a swarm optimizer using a large ensemble of protein decoys [6]. sOPEP2 is the objective function that drives the model building process.
sOPEP (Optimized Potential for Efficient structure Prediction) is expressed as a sum of local, nonbonded and hydrogen-bond (H-bond) terms:
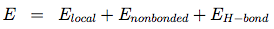
The local potentials are expressed by:
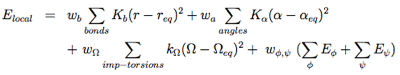
The term Elocal contains force constants associated with changes in bond lengths and bond angles of all particles as well as force constants related to changes in improper torsions of the side-chains and the peptide bonds.
The nonbonded potentials are expressed by:
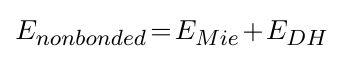
where EMie stands for the use of a Mie formulation instead of the Van der Waals one, and EDH stands for the use of the Debye-Hueckel formalism for interactions between charges.
The Mie potential is expressed as:
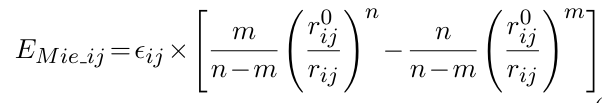
where ![]() is the
potential depth and
is the
potential depth and ![]() is the
position of the potential minimum for interactions between atomic types i and j, and rij is the actual
distance between the beads. More information is available in [6])
is the
position of the potential minimum for interactions between atomic types i and j, and rij is the actual
distance between the beads. More information is available in [6])
The Debye-Hueckel contribution is expressed as:
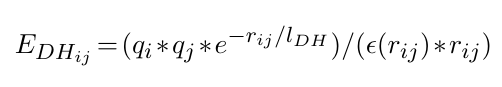
where qi and qj correspond to the charge of particles i and j, j > i+1, respectively. rij is the
distance between the particles, lDH is the Debye length that depends of the ionic strength of the
solvent, and ![]() is the
dielectric constant that depends on the distance between the charges. It is evaluated as:
is the
dielectric constant that depends on the distance between the charges. It is evaluated as:
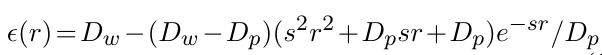
More information is available in [7])
The hydrogen-bonding potential (EH−bond) consists of two-body (EHB1) and four-body (EHB2) terms. Two-body H-bonds are defined by:
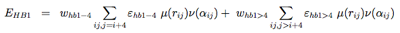
Four-body effects, which represent cooperative energies between hydrogen bonds ij and kl, are defined by:
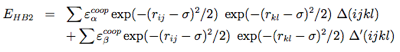
Validation tests using sOPEP2 are presented in [6 and 7].
PEP-FOLD has been tested successfully using various OS / browser combinations, including:
There is presently very few feedback reporting problems with PEP-FOLD:
Mobyle upload button will not work. This spurious behavior has been observed repeatedly. If you encounter such behavior, just perform a full refresh of the result page (press Ctrl+R or press Enter in the url specification field of the browser, or press F5).
As PEP-FOLD4 is a web service available on the RPBS Mobyle portal, it is subject to its terms of use.
Instructions to generate Docker images allowing to run PEP-FOLD4 on your local machine can be found here.
Please note that the access and usage of PEP-FOLD4 through Docker images is subject to this Licence. Key terms of this licence are:
[1] Camproux AC, Gautier R, Tuffery P.
A hidden markov model derived structural alphabet for proteins.
J Mol Biol. 2004 Jun 4;339(3):591-605.
[2] Maupetit J, Derreumaux P, Tuffery P.
A fast and accurate method for large-scale de novo peptide structure prediction.
J Comput Chem. 2010 Mar;31(4):726-38.
[3] Maupetit J, Tuffery P, Derreumaux P.
A coarse-grained protein force field for folding and structure prediction.
Proteins. 2007 Nov 1;69(2):394-408.
[4] Lamiable A, Thevenet P, Tufféry P
A critical assessment of hidden markov model sub-optimal sampling strategies applied to the generation of peptide 3D models.
J Comput Chem. 2016 Aug 5;37(21):2006-16. doi: 10.1002/jcc.24422.
[5] Shen Y, Maupetit J, Derreumaux P, Tufféry P.
Improved PEP-FOLD approach for peptide and miniprotein structure prediction
J. Chem. Theor. Comput. 2014; 10:4745-4758
[6] Binette V, Mousseau N, Tufféry P.
A Generalized Attraction-Repulsion Potential and Revisited Fragment Library Improves PEP-FOLD Peptide Structure Prediction.
J. Chem. Theory Comput. 2022 Apr 12;18(4):2720-2736.
[7] Tufféry P, Derreumaux P.
A refined pH-dependent coarse-grained model for peptide structure prediction in aqueous solution.
Frontiers in Bioinformatics, 2023 3.