PEP-FOLD 3
De novo peptide structure prediction.
De novo peptide structure prediction.
PEP-FOLD is a de novo approach aimed at predicting peptide structures from amino acid sequences. This method, based on structural alphabet SA letters to describe the conformations of four consecutive residues, couples the predicted series of SA letters to a greedy algorithm and a coarse-grained force field.
PEP-FOLD former version available here is based on the greedy strategy can perform 3D modeling for linear peptides up to 36 amino acids, and allows user specified constraints such as disulfide bonds and inter-residue proximities.
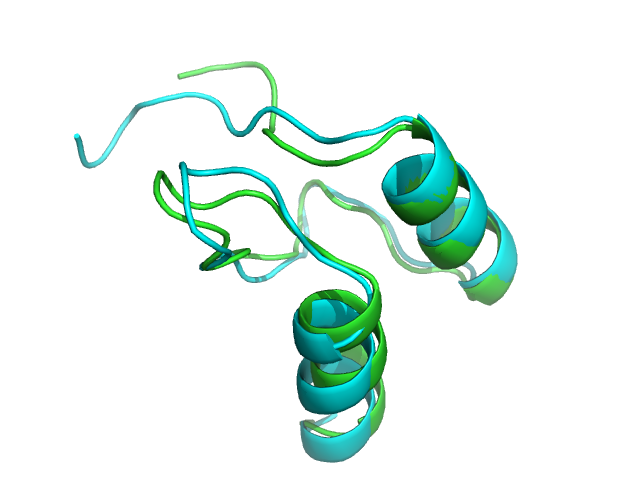
1w4g PEP-FOLD best model (cyan) and experimental (green) conformations. Rigid core BCscore: 0.97 (1 is perfect, 0 is random, -1 is mirror).
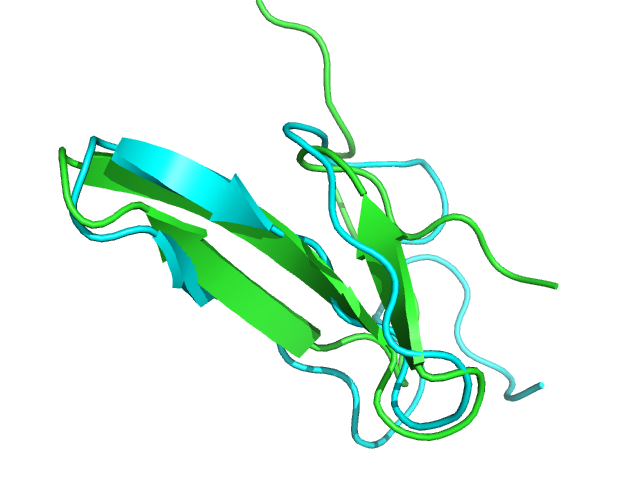
2ysh PEP-FOLD best model (cyan) and native (green) conformations. Rigid core BCscore: 0.89.
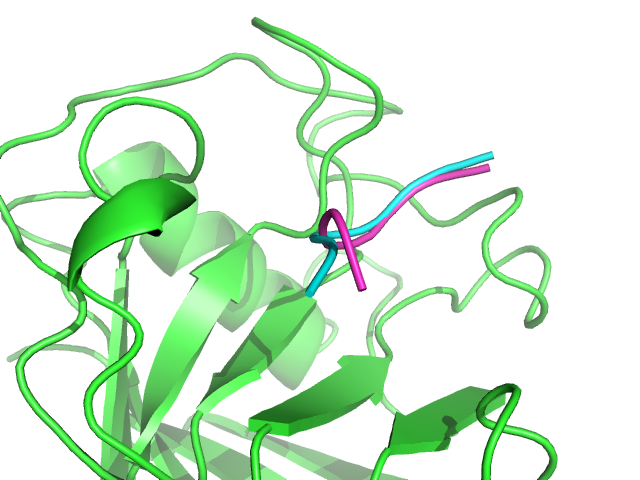
1AWR PEP-FOLD best complex (magenta) and experimental (green/cyan) generated. Peptide RMSd is of 1.34 A.
Access the service through the RPBS Mobyle portal:
When using this services, please cite the following reference:
PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex.
Nucleic Acids Res. 2016 Jul 8;44(W1):W449-54.
Improved PEP-FOLD approach for peptide and miniprotein structure prediction
J. Chem. Theor. Comput. 2014; 10:4745-4758
PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides.
Nucleic Acids Res. 2012. 40, W288-293.

Starting from a single amino acid sequence from 5 to 50 standard amino acids, PEP-FOLD3 runs series of 100 simulations. Each simulation samples a different region of the conformational space. It returns an archive of all the models generated, the detail of the clusters and the best conformation of the 5 best clusters. Note the faster simulation engine makes possible to run PEP-FOLD3 on only one 8 cores machinesn usually still returning models in less than half an hour (this may vary depending on server load).
The PEP-FOLD 3 service accepts information to bias the 3D modeling for user specified regions. User input can impose that some pre-defined local conformations are preserved during modeling, either by providing a former 3D model and indicating which regions are to be propagated to the simulations, or by indicating a list residues for which the PEP-FOLD internal optimal prediction can be considered as accepted. This makes possible iterative modeling in the aim to refine former models, or to geenrate decoy conformations sampling different topologies for instance.
PEP-FOLD3 accepts to fold peptides in the viccinity of a protein receptor, starting from the fuzzy definition of a patch of interaction defined by the user. It is important to understand that the models generated for protein-peptide complexes are generated using a coarse grained representation, which most often must be supplemented by further refinement steps that are not presently included in the PEP-FOLD3 server. During the modeling of protein-peptide interactions, several starting points are generated close to the protein from the user specified patch, then the peptide is grown one residue by one residue in the presence of the protein. Once complete, pepetide structure is refined using a Monte Carlo procedure. The peptide is fully flexible, the protein coordinates are unaffected (rigid).
Presently, PEP-FOLD prediction is limited to amino acid sequence between 5 and 50 residues, although PEP-FOLD has been tested off-line for sizes up to 80 amino acids. For sizes more than 50 amino acids, users can contact the authors. For peptides shorter than 5 amino acids that are usually unstructured, conformational sampling approaches based on molecular dynamics simulations or approaches developped for small compounds should be preferred.
In date of january 2016, PEP-FOLD 3 only accepts the 20 usual amino acids. It will not process peptides with D-amino-acids, or unusual L- amino acids.
Presently, PEP-FOLD3 modeling engine, while able to deal with disulfide bonds, has not shown so far able to perform more efficiently that PEP-FOLD2. PEP-FOLD2 should be used for such peptides. Neither PEP-FOLD2 nor PEP-FOLD3 are able to process circular peptides cyclized by other means than disulfide bonds.
This field is to specify the amino acid sequence of the peptide. The input sequence file must be in FASTA format. The query peptide sequence must contain a string of only the 20 standard amino acids in uppercase, using the 1 letter code (see the pre-configured test example). The size of the input sequence can be as long as 50 amino acids.
A label for the files generated. It MUST be a single word (no spaces, no special characters). It will be used to generate the name of the models available for download.
Once generated, models are clustered to identify groups of similar models. The clusters are then sorted using either the sOPEP energy value, or Apollo [6] predicted TMscore (tm). For peptides up to 36 residues, using sOPEP as a key to sort the clusters will often result in proposing native or near native conformations in the top 5 ranks. For larger sizes, it is preferable to consider the Apollo predicted TM score.
This section correspond to optional additional input to bias model generation.
Input reference structure file must be in PDB format. This file must only contains a single model, with no hetero-residues and have the exact same length (in amino acids) as the query sequence.
The mask specifies which residues in the sequence will have conformational variability. The input sequence must be in FASTA format. It MUST corresponds to the exact same amino acid sequence as that input, but is sensitive to the difference between uppercase and lowercase. The local conformation of residues written using lowercase will not vary, while that of the residues written in uppercase will.
It is important to understand that conformational constraints are expressed in the terms of the structural alphabet (SA) used by PEP-FOLD3 (see the Concepts section for more information). Since the SA considers a peptide as a series of 4 amino acids overlapping by 3, a peptide sequence of L amino acids will be modeled using series of L-3 SA states. Thus, the first 4 amino acids of the sequence correspond to the first fragment and MUST have the same case. The second SA state makes possible to build the 5th amino acid in the structure, etc.
If a reference structure is specified, it will be decoded as a series of SA letters using the Viterbi algorithm. The SA letters associated with lower case residues will be used for 3D modeling.
If no reference structure is specified, an optimal series of SA letters will be predicted, and the SA letters associated with lower case residues will be used for 3D modeling.
This section correspond to optional additional input to generate protein-peptide complexes.
The structure of the receptor must be in PDB format. This file must only contains a single model, with no hetero-residues. Presently, it should contain ONLY ONE CHAIN. Peptide folding onto multimers is not presently supported.
Presently, PEP-FOLD requires the definition of a foreseen patch on receptor surface on which the peptide will bind. The patch is defined as a series of residues of the receptor structure. Given these residues, PEP-FOLD3 will generate candidate starting coordinates for the peptide modeling. Peptide modeling will associate one of these coordinates with one amino acid of the peptide in a stochastic manner. In addition, initial orientation will also be drawn randomly. This results in a very fuzzy definition of the expected interaction patch.
Residues defining the patch must be specified ONE per line. In the PDB format, one amino acid can be unambiguously identified only by specifying the chain label, the residue number, the residue name and the insertion code.
Here, we use the underscore as a delimiter between these fields, using the format:
_CHAIN-ID_RESIDUE-3-LETTERS-CODE _RESIDUE-NUMBER_INSERTION-CODE_
Example:
_A_THR_1__,_A_THR_2__,_A_ILE_7__.
Tests are configured for three different scenari. When selecting one of these, all input data specified in the other input fields will be ignored. Note that the demos are presently tuned to demonstrate PEP-FOLD 3 features only and are not intended for production. Hence they generate a limited number of models.
PEP-FOLD main output consists in models. On-line interactive visualisation and model selection facilities are however proposed.

This section will incrementally provide information about job progression and errors if any. A typical run should produce a report similar to that. Errors related to the input data specified are now also reported in this section.
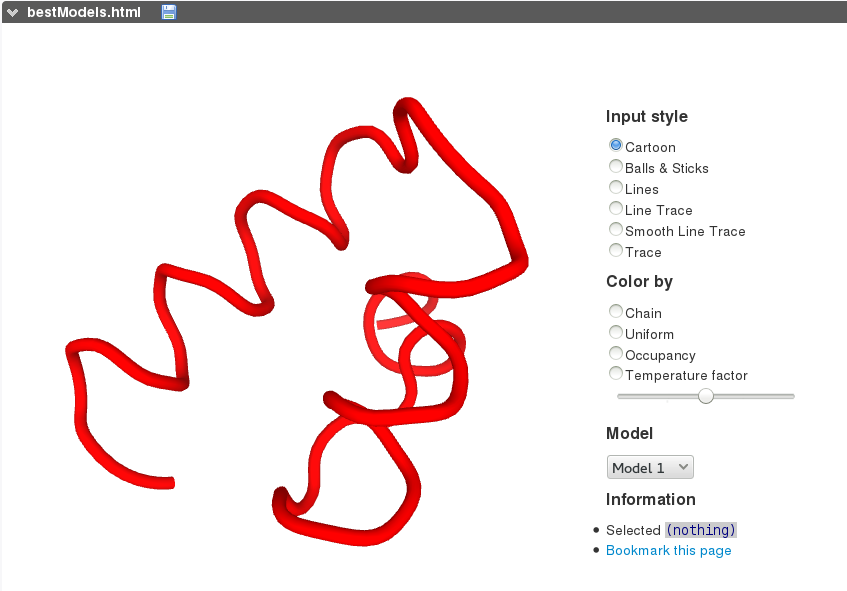
PEP-FOLD3 on-line interactive visualization of the models generated is based on the PV javascript protein viewer . Different representations as well as colouring schemes can be selected. A menu makes possible to select a model among the 10 best models (representatives of the 10 best clusters).

This report is primarily a table file that describes the clusters. For each model generated, up to 10 numbers are reported: avg, gdt, max, q, tm are the scores predicted by Apollo [6], where gdt is the predicted GDT_TS, q is the predicted Qmean score and tm is the predicted TMscore. sOPEP is the coarse grained energy of PEP-FOLD. If a reference file has been specified in input, additional columns correspond to the TMscore and the cRMSd obtained by aligning respectively the model onto the reference using TMalign or a rigid fit procedure minimizing the alpha carbon RMS deviation. The CGCNS and nSS are related to user constraints. CGCNS stands for a Coarse Grained CoNstraint Satisfaction, between 0 and 100, where 100 means all the constraints satisfied. nSS stands for the number of disulfide bond obtained at the all atom representation level (N/A for not appliable).
Additionally, this table gives access to model download at different levels: The archive of all the models (top of table), archives of all models in a cluster - if the cluster contains several models, and each individual models.
The cluster ranks are defined according to their scores (sOPEP or tm) - see the input option "Sort models by". The cluster representatives correspond to the models of the clusters having the best scores, i.e. with the lowest sOPEP energy (resp. highest tm value). They are denoted as "modelx", where x is the rank of the cluster according to the sort key. When a cluster has several models, it is in turn sorted according to the sort key. The first model in the table is the representative, denoted on the form "modelx" and the following models are numbered using the "modelx.y" convention where x is the rank of the cluster and y the rank of the model in the cluster.
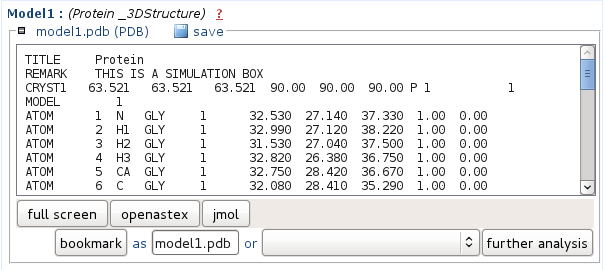
Representatives o the 5 best clusters (according to tm - see Clustering report) predicted structure are provided in PDB format. You can either save the file onto your computer, or view it using JMol. In the Mobyle environment, PDB files can also be piped to other analyses such as the identification of secondary structures using stride or p-sea. For this, select the appropriate method beside the "further analysis" button, then launch it by clicking on "further analysis".
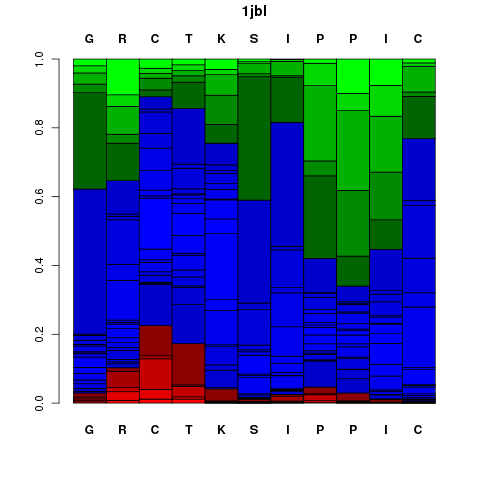
It corresponds to a graphical representation of the probabilities of each Structural Alphabet (SA) - see the Concepts - letter (vertical axis) at each position of the sequence (horizontal axis). Note that SA letters correspond to fragments of 4 residue length. The profile is presented using the following color code: red: helical, green: extended, blue: coil.
This archive contains all the models generated. It is in the unix tar format compressed using gzip.
Once saved on your computer, enter for instance (unix) tar xzf AllModels.tgz to inflate the archive.
As a simple test, you can either choose to:
Copy, paste the following sequence to the "Peptide amino acid sequence" field:
>1egs_A mol:protein length:9 GROES
TKSAGGIVL
or use Mobyle facilities :
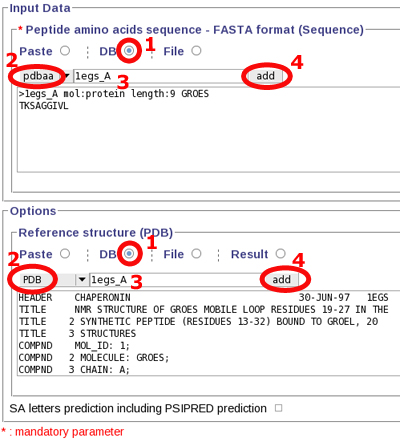
Launch PEP-FOLD prediction, by clicking "Run" at the top of the page.
PEP-FOLD is based on the concept of structural alphabet [1], i.e. an ensemble of elementary prototype conformations able to describe the whole diversity of protein structures.
The OPEP potential helps us to limit the roughness of the peptides energetic landscape, by simplifying side chains representation by a single bead. OPEP v3 parameters are optimized by a genetic algorithm procedure using a large ensemble of protein decoys [3]. OPEP is the objective function that drive the greedy algorithm during the rebuilding process.
OPEP (Optimized Potential for Efficient structure Prediction) version 3 is expressed as a sum of local, nonbonded and hydrogen-bond (H-bond) terms:
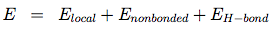
The local potentials are expressed by:
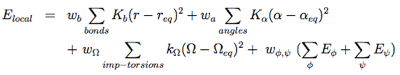
The term Elocal contains force constants associated with changes in bond lengths and bond angles of all particles as well as force constants related to changes in improper torsions of the side-chains and the peptide bonds.
The nonbonded potentials are expressed by:
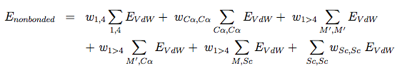
with 1, 4 the 1-4 interactions along each torsional degree of freedom, M ′ the N, C’, O and H main chain atoms, and Sc the side-chain. As seen, we separate short-range from long-range (j > i+4) interactions, and the C alpha atom from the other main chain atoms. For more details on the EVdW potential, see ref [6,5].
The hydrogen-bonding potential (EH−bond) consists of two-body (EHB1) and four-body (EHB2) terms. Two-body H-bonds are defined by:
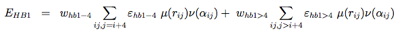
Four-body effects, which represent cooperative energies between hydrogen bonds ij and kl, are defined by:
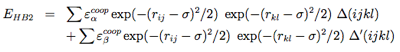
Validation tests for large peptides have been performed on a collection of 56 peptides of size between 25 and 50 amino acids. The results using the former greedy version of the algorithm show PEP-FOLD is able to identify near native or native conformations for 53 among 56 of these peptides, slightly outperforming state of the art de novo approach such as Rosetta [8]. Results using the PEP-FOLD3 engine show identical performance, for slighlty enhanced model quality [7].
PEP-FOLD has been tested successfully using various OS / browser combinations, including:
There is presently very few feedback reporting problems with PEP-FOLD:
In most cases, it is related to a problem of support of WebGL. Please check your browser supports it.
This issue of PV occurs only using the ribbon mode and has been reported to the PV developers.
Mobyle upload button will not work. This spurious behavior has been observed repeatedly. If you encounter such behavior, just perform a full refresh of the result page (press Ctrl+R or press Enter in the url specification field of the browser, or press F5).
[1] Camproux AC, Gautier R, Tuffery P.
A hidden markov model derived structural alphabet for proteins.
J Mol Biol. 2004 Jun 4;339(3):591-605.
[2] Maupetit J, Derreumaux P, Tuffery P.
A fast and accurate method for large-scale de novo peptide structure prediction.
J Comput Chem. 2010 Mar;31(4):726-38.
[3] Maupetit J, Tuffery P, Derreumaux P.
A coarse-grained protein force field for folding and structure prediction.
Proteins. 2007 Nov 1;69(2):394-408.
[4] Kaur H, Garg A, Raghava GP.
PEPstr: a de novo method for tertiary structure prediction of small bioactive peptides.
Protein Pept Lett. 2007;14(7):626-31.
[5] Wang Z, Eickholt J, Cheng J.
APOLLO: a quality assessment service for single and multiple protein models.
Bioinformatics. 2011 27(12):1715-6.
[6] Beaufays J, Lins L, Thomas A, Brasseur R.
In silico predictions of 3D structures of linear and cyclic peptides with natural and non-proteinogenic residues.
J. Pept. Sci. 2011; epub ahead of print.
[7] Lamiable A, Thevenet P, Tufféry P
A critical assessment of HMM taboo sampling strategies applied to the generation of peptide 3D models.
in preparation.
[8] Shen Y, Maupetit J, Derreumaux P, Tufféry P.
Improved PEP-FOLD approach for peptide and miniprotein structure prediction
J. Chem. Theor. Comput. 2014; 10:4745-4758