
DockSurf software runs as a batch job with predetermined inputs to process computations and analyze output data. Input protein 3D coordinates must be exclusively in the protein databank format[1], which is - by far - the most used file format. DockSurf utilizes the free software Gnuplot[2] to generate 3D contour plot images in png format.
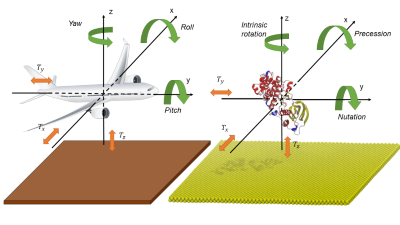
Docksurf tackle the problem of finding the optimal interaction of a protein on an inorganic surface as a molecular docking question: the optimal interaction of a protein on an inorganic surface can be translated by the finding of the best plane on a protein molecular surface. Proteins are considered to be rigid and the process of exploring their structures can be likened to an airplane flying above the ground: an airplane moves through three translations along the x, y, and z axis (Tx, Ty, and Tz) and rotations are defined around these axes.
In the context of protein/surface interactions, it is possible to eliminate certain degrees of freedom that describe the movement of a rigid-body, such as a protein. By considering the mineral surface to be flat and uniform, the Tx and Ty translations, along with intrinsic rotation, can be eliminated. Thus, only the Tz translation and precession and nutation rotations are needed to describe the structural organization above the mineral surface. Docksurf use an astutely defined scoring function to consider only two rotations allowing an exhaustive exploration of these coordinates, providing a cartography of the protein/surface interaction.
The Docksurf software quickly provides a qualitative ranking of the association energy. To this extent, a specific potential, inspired from the work of Derjaguin, Landau, Verwey, and Overbeekhas (DLVO)[3] and their further improvement[4], has been designed in this work. This potential was parameterized with QM and MM computations by investigating nine small molecules representing the protein diversity.
The most striking and specific result of the DockSurf software is to propose a mapping of the interaction between a protein and an inorganic surface according to the orientation of the biomolecule. To let the user choosing wisely the interaction structure(s) of its protein structure, the software produces 5 distinct mappings:
DockSurf software systematically generates the maps with the 5 above mentioned scorings and creates the protein complexes with the inorganic surface for the number of selected structures, as distinct files (in pdb format). The authors of this work strongly suggest that users systematically compare the DGQM and DGMM generated maps in order to confirm an orientation.
It is worth noting that despite their names, and like any molecular docking software, generated scorings are far from being comparable to real free energy computations.
[1] Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E. The Protein Data Bank. Nucleic Acids Res 2000, 28 (1), 235–242. https://doi.org/10.1093/nar/28.1.235.
[2] Williams, T.; Kelley, C. Gnuplot: an interactive plotting program. http://gnuplot.info/ (accessed 2022-07-25)
[3] Overbeek, J. T. G. Recent Developments in the Understanding of Colloid Stability. Journal of Colloid and Interface Science 1977, 58 (2), 15.
[4] Vellore, N. A.; Yancey, J. A.; Collier, G.; Latour, R. A.; Stuart, S. J. Assessment of the Transferability of a Protein Force Field for the Simulation of Peptide-Surface Interactions. Langmuir 2010, 26 (10), 7396–7404. https://doi.org/10.1021/la904415d.