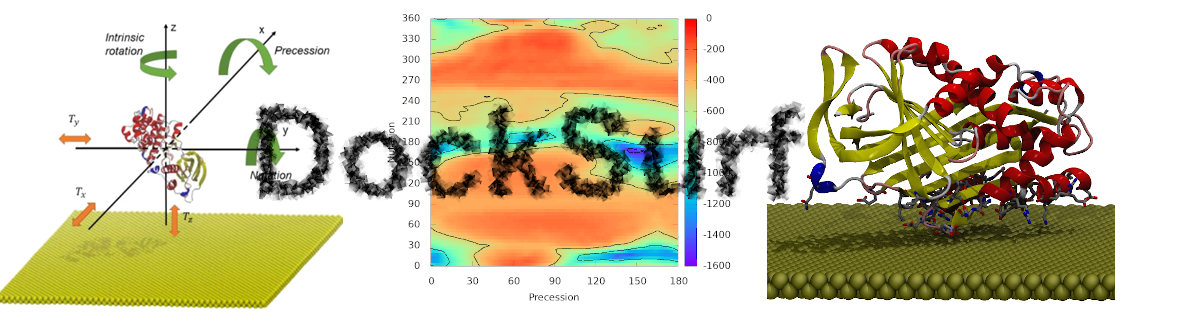
The elucidation of structural interfaces between proteins and inorganic surfaces is a crucial aspect of bionanotechnology development. Despite its significance, the interfacial structures between proteins and metallic surfaces have yet to be fully understood, and the lack of experimental investigation has impeded the development of many devices. To overcome this limitation, we suggest considering the generation of protein/surface structure as a molecular docking problem. To this extent the DockSurf software aims to quickly propose reliable protein/surface structures through conformational exploration with Euler's angles, which provide a cartography instead of a unique structure. This software is freely available and implemented in the RPBS platform to facilitate widespread access to the scientific community.