InterEvDock2
A docking server to predict the structure of protein-protein interactions using evolutionary information.
A docking server to predict the structure of protein-protein interactions using evolutionary information.
The InterEvDock2 service is integrated in the RPBS Mobyle Portal.
This website is free and there is no login requirement. Note that for homology modeling, using this service starting from sequences is allowed only for non-commercial users (non-profit/academic/governement research groups). Commercial users are asked to submit their own models.

Two protein structures (or structural models generated on the fly from input sequences) and their respective multiple sequence alignments are used to predict binding modes through a free docking procedure.
The structural modelling of protein-protein interactions is key in understanding how cell machineries assemble and cross-talk with each other. When homologous sequences are available for both protein partners, it is very useful to rely on structures and multiple sequence alignments to identify binding interfaces. InterEvDock2 is a server for protein docking running the InterEvScore potential specifically designed to integrate evolutionary information in the docking process. The InterEvScore potential was developed for heteromeric protein interfaces and combines a residue-based multi-body statistical potential with evolutionary information derived from the multiple sequence alignments of each partner in the complex. In the InterEvDock2 server, the systematic docking search is performed using the FRODOCK2 program [1] and the resulting models are re-scored with InterEvScore [2] together with the SOAP_PP atom-based statistical potential [3] found to increase the confidence of the predictions.
InterEvDock2 is an update of InterEvDock [4] that can handle protein sequences as inputs, and not only protein 3D structures. When a sequence is provided by the user, a comparative modeling step based on an automatic template search protocol builds models for the individual protein partners, prior to docking. In InterEvDock2, in case the user has biological input such as a position that is known to be involved in the interface between the two protein partners, constraints can be specified for use in the docking procedure. This can be crucial to ensure that all available biologically relevant information is used for InterEvDock2 predictions. In addition, InterEvDock2 implements the possibility to submit structures of oligomers as input to the free docking. Such an option is generally complicated in co-evolution analyses since the joint MSAs have to be generated for every chain of an oligomer. This process is now fully automatized in InterEvDock2.
When using this service, please cite the following references:
InterEvDock2: an expanded server for protein docking using evolutionary and biological information from homology models and multimeric inputs.
Nucleic Acids Res. 2018 Jul 2;46(W1):W408-16.
InterEvDock: A docking server to predict the structure of protein-protein interactions using evolutionary information.
Nucleic Acids Res. 2016 Jul 8;44(W1):W542-9.
InterEvScore: a novel coarse-grained interface scoring function using a multi-body statistical potential coupled to evolution.
Bioinformatics. 2013 29(14):1742-9.
Please, cite also the FRODOCK2 program which is used for the rigid-body docking step:
FRODOCK 2.0: fast protein-protein docking server.
Bioinformatics. 2016;32(15):2386-8.
Using the results of SOAP_PP, please cite :
Optimized atomic statistical potentials: assessment of protein interfaces and loops.
Bioinformatics. 2013;29(24):3158-66.
Using the evolutionary conservation results obtained using Rate4Site (mapped onto all visualized models in the PV applet and written into the b-factor field of the PDB files provided for all models in the results zip archive) please cite:
Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues.
Bioinformatics. 2002; 18 Suppl 1:S71-77.
Using the comparative modeling protocol based on RosettaCM (i.e. if your input consists in one or two sequences), please cite:
High resolution comparative modeling with RosettaCM.
Structure. 2013; 21(10):10.
Using the automatic template search (i.e. if your input consists in one or two sequences and you did not specify a template), please cite:
HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment.
Nat Methods. 2011;9(2):173-5.
Protein homology detection by HMM-HMM comparison.
Bioinformatics. 2005; 21(7):951-60.
InterEvDock2 takes as input either the structures of two protein partners to be docked (experimental or modelled structures, possibly multimeric), or their sequences.
Given the sequences, the server runs several steps to generate a model of the structures:
Given the structures (either input by the user or modeled from input sequences), the server runs several steps to propose a selection of 10 most likely models for each score (InterEvScore, SOAP_PP, FRODOCK scores) as well as 10 consensus models and 5 most likely interface residues on each protein:
The docking server implements 3 major methods:
FRODOCK2 (developed in Pablo Chacón's lab [1])., a rigid-body docking method which combines a search algorithm based on spherical harmonics, an energy-based scoring function including van der Waals, electrostatics and desolvation terms and a complementary knowledge-based potential. FRODOCK2 has competitive success rates and efficiency when challenged on Weng's Benchmark v4[9].
InterEvScore , a scoring function combining a residue-based statistical potential including both two- and three-body statistical potentials with the scoring of interface contacts inferred from multiple sequence alignments. This way of integrating evolutionary information was found significantly superior to solely accounting for conserved positions. The server version includes the mode in which InterEvScore uses evolutionary information only for residues belonging to apolar patches as described in [2] (developed in Guerois' lab see InterEvScore [2]).
SOAP_PP , an atom-based statistical potential dedicated to protein-protein interactions derived from a general Bayesian framework for inferring statistically optimized atomic potentials (SOAP) in which the reference state is replaced with data-driven ‘recovery’ functions. Relative orientation between two covalent bonds instead of a simple distance between two atoms contribute to capture orientation-dependent interactions such as hydrogen bonds. (developed in Andrej Sali's lab [3])
InterEvDock is a method for generating complex between two protein structures modelled as rigid-body subunits. Selected models may have some clashes that can be released upon relaxation of the models.
InterEvDock supposes the interaction between the two submitted subunits has been experimentally validated. It is not designed to predict neither the likelihood nor the strength of the interaction.
Large conformational changes upon binding are generally not well predicted.
The simplest input consists in two fields to specify either the sequence or the 3D structures of the proteins to be docked.
Protein A structure or sequence: Fill one of the two fields. Warning: if both fields are filled, only the protein structure will be taken into account.
These controls address two different points:
Two controls are available:

Breakpoints:

A list of constraints can be optionally specified. Each constraint should be either a position from protein A or protein B or a pair of positions, one from protein A and one from protein B. Each constraint must contain at least one colon ":" used to specify which of the two partners the constraint applies to (before the colon for the first partner, after the colon for the second partner. e.g.
11A:
if we want residue at position 11 on chain A in protein A to be at the interface;
:20B
if we want residue at position 20 on chain B in protein B to be at the interface;
11A:20B
to enforce a contact between position 11 on chain A in protein A and position 20 on chain B in protein B). Positions are numbered according to the PDB numbering if the input structure was provided and according to sequential numbering of the FASTA sequence if only the input sequence was provided. In the latter case, the chain name should be A by default. Several constraints can be specified, separated by whitespace (space, tabulation or newline). A distance can be optionally specified for each constraint, for instance
11A::7
for a distance of 7 Å on position 11A (note the two colon “:” separators) and
11A:20B:7
for a distance of 7 Å between 11A and 20B. The default distance is 8 Å for single residues and 11 Å for pairs. These constraints will be taken into account in the docking process to keep only models having this position or pair of positions at the complex interface. Constraints will be checked prior to applying and constraints involving residues not present or buried in the structural model will be excluded. Note that when several constraints are provided, they are considered cumulative ("AND" not "OR") i.e. docking models will be filtered to retain only solutions that verify all constraints.
Positions are numbered according to the PDB numbering if the input structure was provided and according to sequential numbering of the FASTA sequence if only the input sequence was provided. In the latter case, the chain name should be A by default. Several constraints can be specified, separated by whitespace (space, tabulation or newline). A distance can be optionally specified for each constraint, for instance 11A:7 for a distance of 7 Å on position 11A and 11A:20B:7 for a distance of 7 Å between 11A and 20B. The default distance is 8 Å for single residues and 11 Å for pairs. These constraints will be taken into account in the docking process to keep only models having this position or pair of positions at the complex interface. Constraints will be checked prior to applying and constraints involving residues not present or buried in the structural model will be excluded. Note that when several constraints are provided, they are considered cumulative ("AND" not "OR") i.e. docking models will be filtered to retain only solutions that verify all constraints.
In order to avoid round-trips outside the browser, it is possible to use the on-line NGL Viewer viewer to quickly identify residues.

Two fields can optionally be filled if the user would like a specific template to be used in the modeling.
Same as above for the second protein partner.
Structural models are generated following the sequence-template alignment calculated by the server or provided by users in the dedicated frame.

In standard conditions, insertions (loops) are modeled up to a length of 14 residues. Beyond this limit residues are not modeled and a break in the chain is created. Long loops might be detrimental for the validity of the subsequent rigid-body docking so that users can tune the length of the loops and reduce this threshold parameter. Similarly, tails at the N-terminus and C-terminus are generally not modeled if they are not observed in the template PDB. Users can decide to model them up to a certain number of the residues by changing the parameters. In the sequence-template alignment frame, N-ter and C-ter tail residues will correspond to residues that are aligned with gaps in the extremities of the alignment.
Two fields can be optionally filled to specify the multiple sequence alignments associated with each 3D structure. The first sequence in each alignments should match as closely as possible the sequence of the corresponding PDB file (except gaps represented by '-'). The two alignments must contain sequences from the same set of species, appearing in the exact same order. If the multiple sequence alignments are not provided, they will be automatically generated by the InterEvDock server and will be provided in the results to facilitate potential re-submission.
Accessible from InterEvDock2 page by setting "Yes" on the Demonstration Mode. By setting this option to "Yes", InterEvDock2 will load a pre-configured test case where the complex between the STAS domain and the acyl carrier protein in bacterium Hahella chejuensis is modeled.
There are four versions of the Demonstration Mode, starting from input sequences only.
The results can be compared with the coordinates of the reference PDB complex between the same proteins in Escherichia coli (PDB:3NY7). By switching on the demonstration mode, other input data specified in the other fields will be ignored.
Progress report

This section will incrementally provide information about job progression and errors if any. A typical run should produce a report similar to the one shown above. Errors related to the input data specified are also reported in this field.
The results related to template identication are divided in four sections.




PDB files of the models generated for protein A and/or protein B are presented on the form:

It is possible to visualize interactively the structure by clicking on one of the PV or NGL button located at the bottom of the window. Using PV, moving the mouse cursor, residue names and numbers are displayed in the selected text field, which can be used to define constraints for the docking. Using NGL, activating the label item will trigger the display of residue labels.


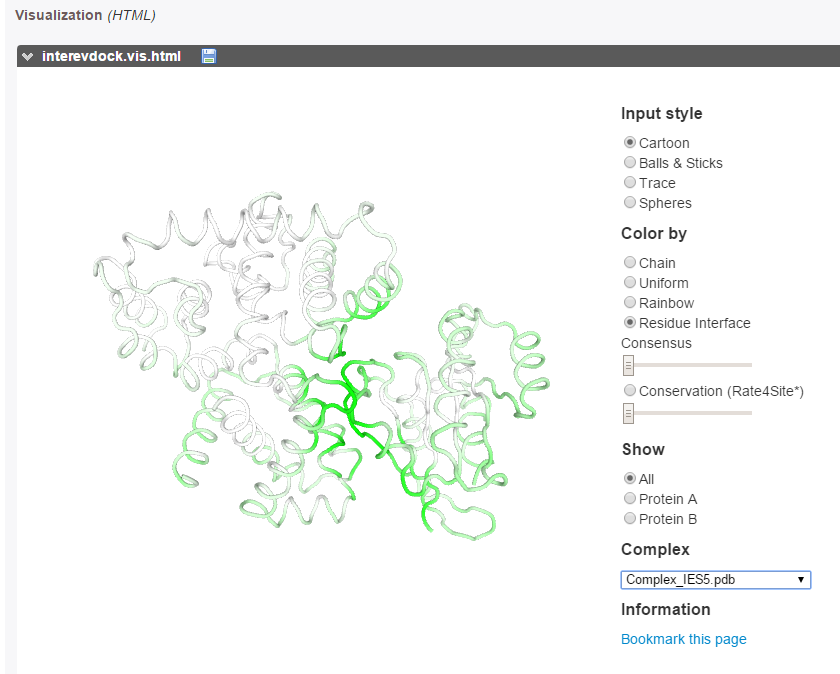
The best models can be explored using the PV applet.
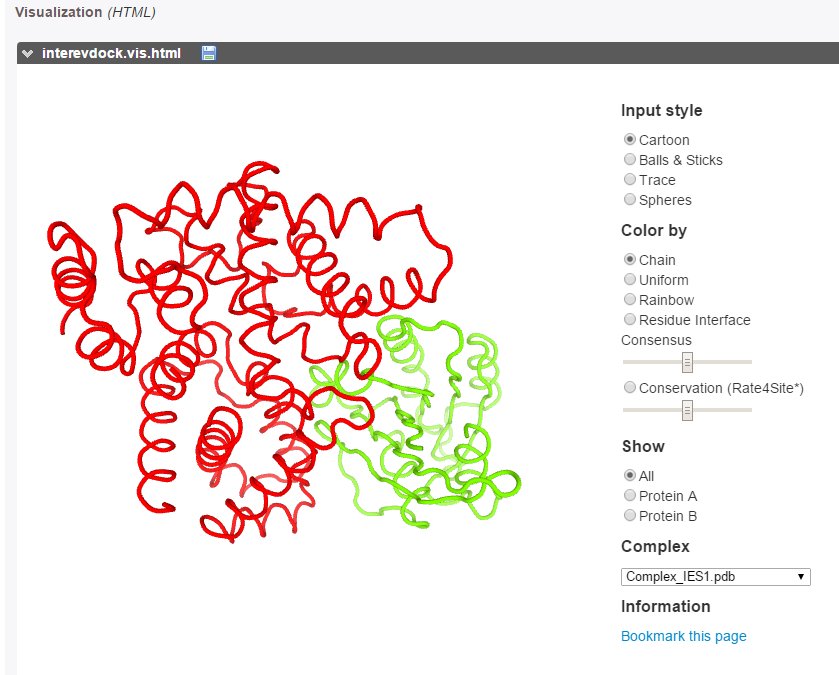
The predicted interface residues can also be visualized on both proteins as a color gradient(from green to white for high to low probability to be at the interface).
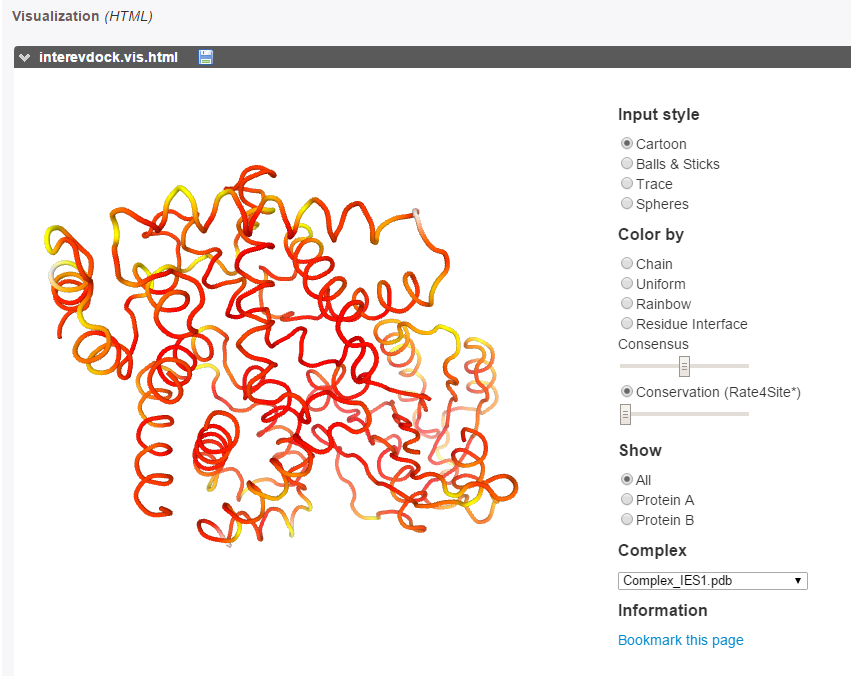
The evolutionary conservation of each partner (calculated with Rate4Site [10]) can be visualized on both partners as a color gradient, from red (more conserved) to white (more diverse) through yellow (mild conservation).
A PyMOL script provided in the results zip archive for each run automatically loads the 30 models from the results zip file and colors them by interface residue consensus and evolutionary conservation.

The InterEvScores, SOAP_PP and frodock scores are reported for the 10 best complexes.

If constraints were specified in input, information about each constraint satisfaction is presented as a table.
The result page of this demo executed with a breakpoint after template search can be accessed here and the result page of the demo executed with a breakpoint after modeling can be accessed here.
The result page of this demo without constraint can be accessed here and the result page of the demo with constraint 69A can be accessed here.

Example of the best ranked InterEvDock2 model using constraint 69A for the H. chejuensis complex between the STAS domain (in shades of yellow and red) and the ACP protein (in shades of blue), compared to the reference E.coli complex (PDB 3NY7, in dark grey).
Starting from input sequences only, InterEvDock2 builds models of the two chains and performs the docking procedure. The results can be compared with the coordinates of the reference PDB complex between the same proteins in Escherichia coli (PDB:3NY7). The STAS domain of H. chejuensis has 43% sequence identity with the STAS domain of E. coli and the ACP has 79% sequence identity. When using no constraint, the H. chejuensis STAS/ACP model ranked as top 6 in the top 10 consensus with an iRMSD of 2.3 Å ("Acceptable" quality according to the CAPRI criteria) compared to reference complex 3NY7. When using a constraint on position 69A involved in the interface, the best model (top 1 in the top 10 consensus) has an iRMSD of 3.95 Å ("Acceptable" quality according to the CAPRI criteria).
In the CAPRI30 session in which our group performed very well [11], targets of CASP11 crystallizing as homo-oligomers were proposed as CAPRI targets. Using the same strategy as implemented in the InterEvDock pipeline (rigid-body docking followed by scoring with multiple scores including InterEvScore), we submitted the model with the lowest interface RMSD (3.5 Å) for target T72. In case of target T72, template-based modelling led to misleading assembly prediction (right figure). Accordingly, at low sequence identity, assembly modes can substantially differ between remote homologs.
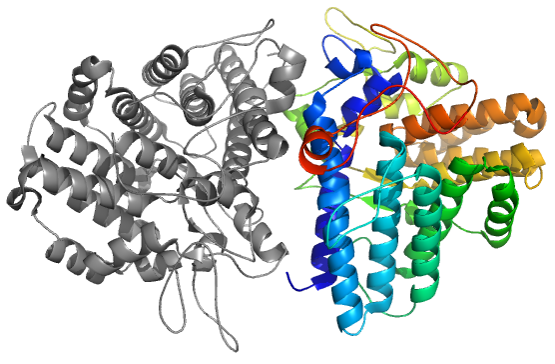
Xray structure of CASP11 target T0770 (4Q69), CAPRI30 Target72
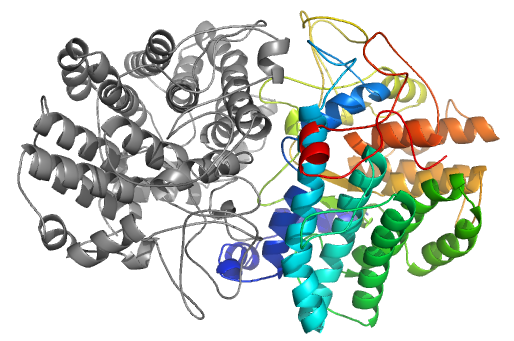
Structural model generated with correct orientation rated as "Acceptable" by CAPRI with interface RMSD of 3.5 Å obtained following a free docking procedure using the InterEvDock pipeline
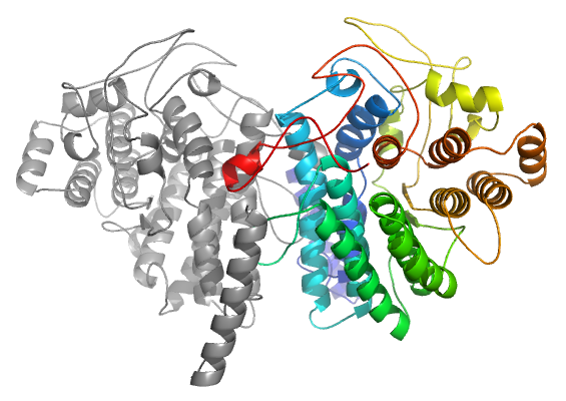
Incorrect model resulting from a simple template-based modelling based on pdb 3MX3 (seq. Id 20 %)
InterEvScore achieves significant improvement over traditional scoring functions on the 54 test cases from Weng's docking benchmark v4 with available coupled multiple sequence alignments and near-native decoys [2]. The use of evolution increased the quality of the prediction and was never detrimental in the discrimination of near-native interfaces, even though the number of sequences in the coupled alignments could be limited (between 10 and 100 species with an average of 35). In addition, we did not find the scoring improvement on inclusion of evolutionary data to be limited to certain categories of complexes (except for antibody-antigen complexes).
Performance of InterEvDock2 was assessed on 812 complexes from the PPI4DOCK database [12] for which the structures of the free proteins (unbound) can be modeled and for which evolutionary information could be retrieved [2]. A table of all 812 benchmark cases is available here.
The InterEvDock2 server predicts an "Acceptable" or better solution in the consensus top10 for 239 out of 812 test cases (29%). As expected, this top10 success rate decreases with increasing difficulty of the docking cases: 43% for very easy targets, 30% for easy targets, 11% for hard targets and 5% for very hard targets according to PPI4DOCK difficulty classification.
The InterEvDock2 server also predicts residues making contacts at the interface of a complex based on the analysis of all the interfaces of the top10 decoys for all three scores (30 models). In 91 % of the 812 test cases, at least one residue out of 10 was correctly predicted as present at the interface, providing very useful hints to guide mutagenesis experiments to disrupt a complex of interest. Of note, there is little decrease in precision from the rigid-body to the difficult cases (success rate is 92% for very easy targets, 90% for easy targets, 87% for hard targets and 100% for very hard targets according to PPI4DOCK difficulty classification). Predictions of the InterEvDock2 server can thus also be used as a prior to constrain more thorough docking simulations requiring flexibility in order to model the correct orientation between two binding partners. In that perspective, in 51% of the cases, at least one correct residue is predicted on both sides of the interface (59% for very easy targets, 53% for easy targets, 33% for hard targets and 41 for very hard targets according to PPI4DOCK difficulty classification). When considering only the top 1 predicted residue on each chain, at least one of the two predicted residues is correct in 75% of the cases and both are correct in 34% of the cases, highlighting the practical value of InterEvDock2 residue prediction. All those results are significantly higher than a reference interval given by random selection of residues on the surface of the protein.
FRODOCK 2.0: fast protein-protein docking server.
Bioinformatics. 2016; 32(15):2386-8.
InterEvScore: a novel coarse-grained interface scoring function using a multi-body statistical potential coupled to evolution.
Bioinformatics 2013; 29 (14):1742–1749.
Optimized atomic statistical potentials: assessment of protein interfaces and loops.
Bioinformatics. 2013; 29(24):3158-66.
InterEvDock: A docking server to predict the structure of protein-protein interactions using evolutionary information.
Nucleic Acids Res. 2016; 44(W1):W542-9.
BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences.
FEMS Microbiol Lett. 1999;174(2):247-50.
HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment.
Nat Methods. 2011;9(2):173-5.
Protein homology detection by HMM-HMM comparison.
Bioinformatics. 2005; 21(7):951-60.
High resolution comparative modeling with RosettaCM.
Structure. 2013; 21(10):10.
Protein-protein docking benchmark version 4.0.
Proteins. 2010; 78(15):3111-4.
Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues.
Bioinformatics. 2002; 18 Suppl 1:S71-77.
Table CAPRI30: Ranking by number or INTERFACES for which at least one 'Acceptable' solution was obtained.
In : Presentation from the CAPRI30 Cancun meeting
PPI4DOCK: large scale assessment of the use of homology models in free docking over more than 1000 realistic targets.
Bioinformatics. 2016; 32(24):3760-3767.
This work was supported by the French Infrastructure for Integrated Structural Biology (FRISBI) [ANR-10-INSB-05-01]; ANR-IAB-2011-BIP:BIP [ANR-10-BINF-0003]; IFB [ANR-11-INBS-0013]; CHIPSET [ANR-15-CE11-0008-01]. PPI4DOCK benchmarking was done through granted access to the HPC resources of CCRT under the allocations 2015-7078, 2016-7078 and 2017-7078 by GENCI (Grand Equipement National de Calcul Intensif).
![[11]](images/CAPRI30.png){kind=link}