Overview
In silico assessment of protein receptor interactions with small ligands is now part of the standard pipeline for drug discovery, and numerous tools and protocols have been developed to this aim. The SeamDock on-line service integrates different docking tools in a common framework that makes possible to undergo ligand global and/or local docking and a hierarchical approach combining the two for easy interaction site identification. This service does not require advanced computer knowledge and it works without installation of any programs with the exception of a common web browser. The use of the seamless library linking the RPBS calculation server to the user's web page, allows the user to navigate smoothly and interactively on the SeamDock web page. A major effort has been put into the 3D visualization of ligand, receptor, and docking poses and their interactions with the receptor. The advanced visualization features combined with the seamless library allow a user to share with an unlimited number of collaborators, a docking session and its full visualization states. As a result, SeamDock can be seen as a free, simple, didactic, evolving on-line docking resource best suited for education and training.
Access the service through the RPBS Web Portal
This website is free and open to all users and there is no login requirement.
Pre-calculated result from the literature
When using this service, please cite the following reference:
Murail S, de Vries S, Rey J, Moroy G & Tufféry P.
SeamDock: An Interactive and Collaborative Online Docking Resource to Assist Small Compound Molecular Docking.
Frontiers in Molecular Biosciences (2021). https://doi.org/10.3389/fmolb.2021.716466
Tufféry P & Murail S.
Docking_py, a python library for ligand protein docking.
Zenodo (2020), http://doi.org/10.5281/zenodo.4506970.
WARNING: This service is under developpment. Use it with caution.
Usage
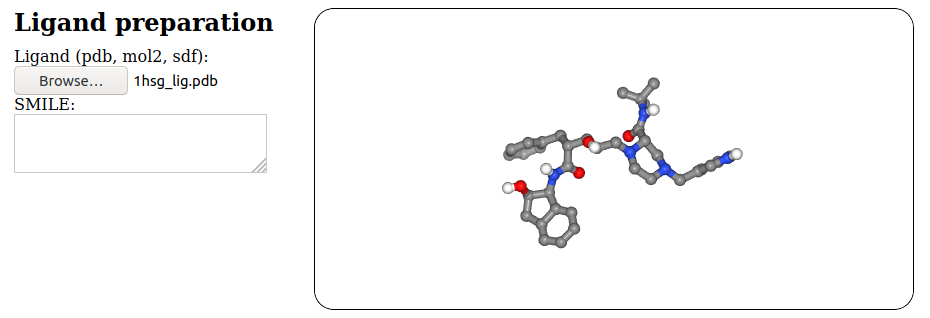
The main input for SeamDock is simply a ligand and a receptor.
The ligand input can consist of:
- An input file, as
.pdb, .mol2 or .sdf files
- A Simplified Molecular-Input Line-Entry System (SMILES) string eg.
"CCO" for ethanol
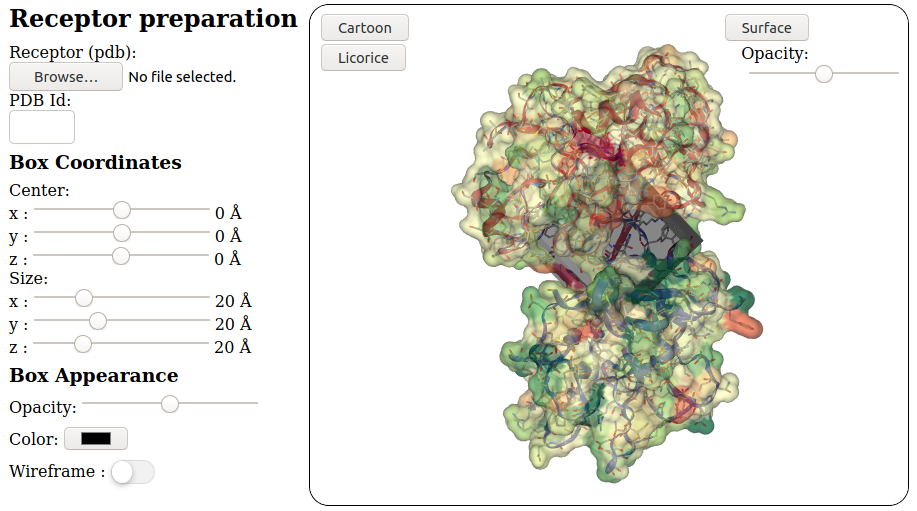
The receptor input can consist of:
- A
.pdb, file
- A PDB accession code. eg.
"3EAM" for GLIC protein
Once the receptor has been loaded and treated, user should define the docking box parameters:
- 3D coordinates of the docking box
- Size (x, y, z) of the docking box
The following receptor and box appearance can be set by the user:
- Receptor
- Receptor Cartoon vizualisation on/off (default: on)
- Receptor Licorice vizualisation on/off (default: on)
- Receptor Surface on/off (default: on)
- Receptor Surface oppacity (default: 50%)
- Docking box
- Wireframe / Surface Box vizualisation switch (default: Surface)
- Color (default: black)
- Box oppacity (default: 50%)
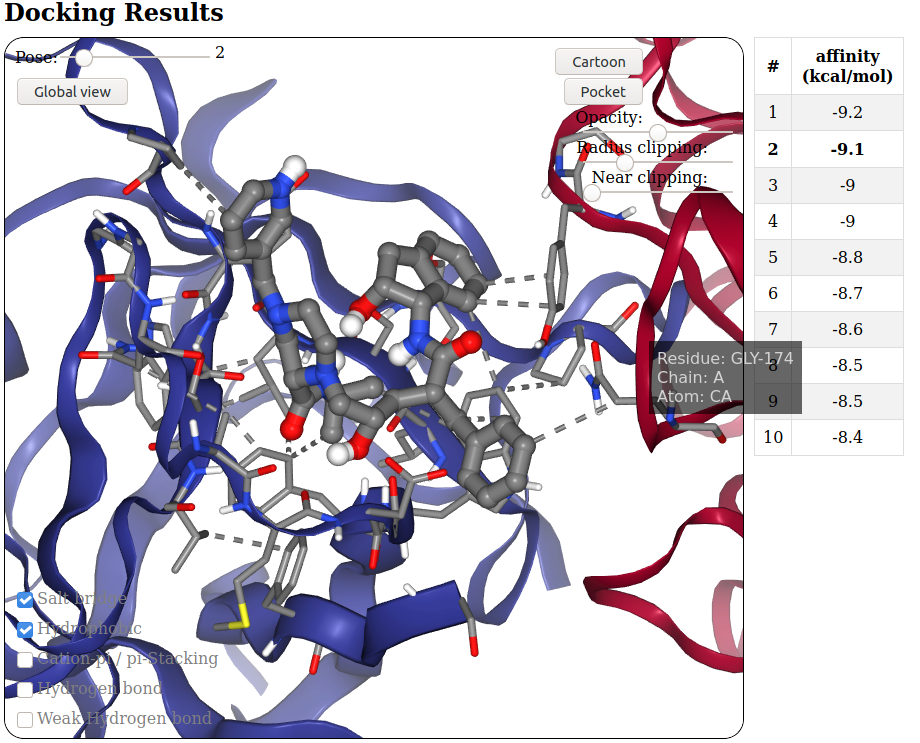
Usage: output
This section will incrementally provide information about job progression and errors, if any.
A typical run should produce a progress report similar to the following:
Interpreting the results
TO DO !
Browser compatibility
| OS |
Version |
Chrome |
Firefox |
Microsoft Edge |
Safari |
| Linux |
Ubuntu 20 |
87.0 |
84.0 |
n/a |
n/a |
| MacOS |
Mojave |
87.0 |
84.0 |
n/a |
12.0 ⚠ |
| Windows |
10 |
87.0 |
84.0 |
87.0 |
n/a |
Project history
- 2021, April: User interface of the web server.
- 2020, December: Docking_py Code uploaded in the Zenodo (DOI: 10.5281/zenodo.4506970).
References
[1] Tufféry P & Murail S.
Docking_py, a python library for ligand protein docking.
Zenodo (2020), http://doi.org/10.5281/zenodo.4506970.
[2] Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S. & Olson, A. J.
Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity.
J. Computational Chemistry (2009), 19: 1639-1662.
[3] Trott, O. & Olson, A. J.
AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading.
J. Computational Chemistry (2010), 31: 455-461.