PEP-Cyclizer
A large scale structure mining approach to assist the design of peptide head-to-tail cylization.
A large scale structure mining approach to assist the design of peptide head-to-tail cylization.
PEP-Cyclizer is a tool to assist the design of head-to-tail peptide cyclization, a well known strategy to enhance peptide resistance to enzymatic degradation and thus peptide bioavailability.
Access the service through the RPBS Mobyle portal:
When using this service, please cite the following reference:
PEP-Cyclizer: a service to assist peptide cyclization through a 3D structure mining approach. in preparation

Although cyclic peptides as short as 4 amino acids exist, PEP-Cyclizer paradigma is more oriented towards the cyclization of bioactive peptide formerly characterized, and consequently often peptides of larger sizes. In practice, PEP-Cyclizer will not start from un-cyclized peptides of size less than 8 amino-acids, due to the requirement to perform the PDB mining (see farther).
PEP-Cyclizer only accepts un-cyclized peptides made of the 20 usual amino acids. It will not process un-cylized peptides with D- amino-acids, or unusual L- amino acids.
When guessing for candidate sequences, it will only propose sequences made of the 20 usual L- amino acids.
It is possible to undergo the head-to-tail cyclization of peptides containing disulfde bonds. However PEP-Cylizer may not account correctly for other kinds of peptide internal cyclizations prior to head-to tail cyclization. For instance, the design of bicyclic peptides is out of the scope of PEP-Cyclizer.
This field is to specify the coordinates of the template peptide. The input file must be in PDB format. When multiple models are provided, only the first one will be considered. It must contain only the 20 standard amino acids. The size of the input template can be as long as 50 amino acids, but must be longer than 8 amino acids. Note that residue numbers will be changed for the ouptut, the first amino-acid will assigned number 1.
This field is to specify the sequence of a candidate linker. Its size may vary between 2 and 10 amino acids. Its sequence must be provided using the 1 letter amino acid code. Filling this field, PEP-Cyclizer will generate 3D models for the head-to-tail cyclized peptide.
This field is to specify the size of a candidate linker. Its size may vary between 2 and 10 amino acids. Filling this field (default), PEP-Cyclizer will infer information from the PDB fragments of the specified linker size matching the head-to-tail closure condition.
It is often undesirable to perform head-to-tail cyclization with amino acids likely to interfere with peptide bioactivity. For this reason, only a limited subset of amino acids is most often used. This field makes possible to specify constraints on the amino acid types at each position. Information for each position in the linker sequence MUST be provided. the ':' symbol is used as a delimiter.
1:F:G:P indicates that at linker position 1, only PHE, GLY and PRO are preferred.
1: indicates that at linker position 1, no preference is specified, meaning all 20 standard amino acids are allowed.
By default the form is filled using 1:A:G, i.e. indicating a preference for ALA and GLY.
Tests are configured for two different scenari. When selecting one of these, all input data specified in the other input fields will be ignored.
PEP-FOLD main output consists in models. On-line interactive visualisation and model selection facilities are however proposed.

This section will incrementally provide information about job progression and errors if any. A typical run should produce a report similar to that. Errors related to the input data specified are now also reported in this section.
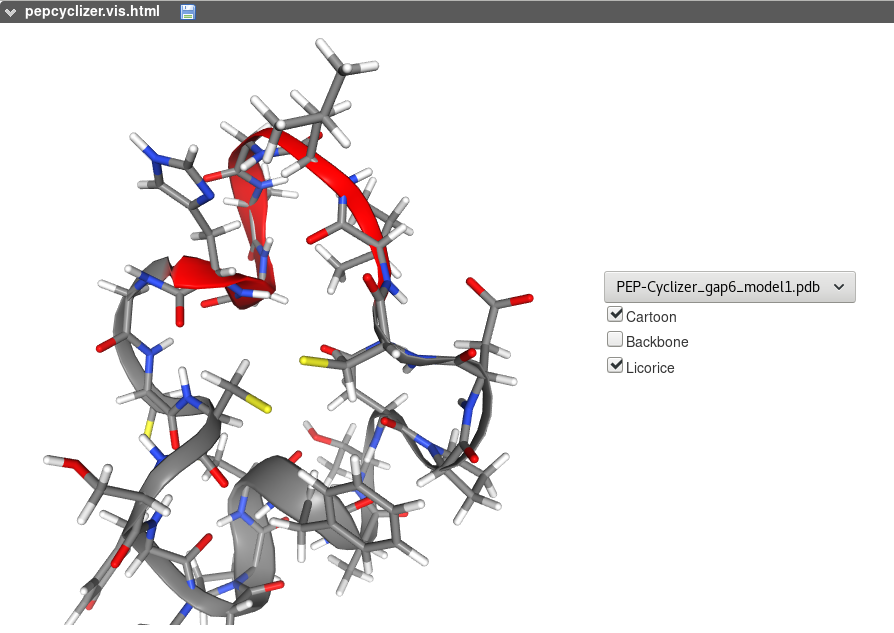
PEP-Cyclizer on-line interactive visualization of the models generated is based on the NGL . Different representations can be selected. A menu makes it possible to select a model among the 30 best models (representatives of the 30 best clusters), or to visualize them together.
This report is a display of the variability of the amino acids observed over the candidate sequences. Red amino acids correspond to the amino acids. We remind that, due to the weak probability to find sequences matching the sequence constraints at all positions, we select, among the candidates matching the closure geometry constraints, sequences that match at least 50% constraints, which explains the occurrence of other amino acids at each position (in gray).
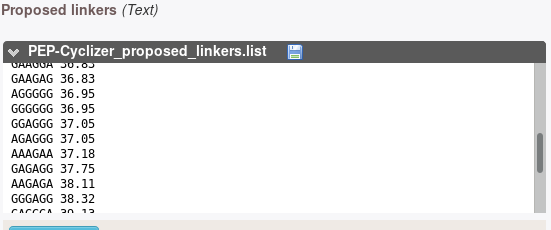
This section presents sequences drawn using the forward backtrack algorithm on the model fit over the data. Each sequence is associated with a score that corresponds to -log(L), where L is the likelihood of the sequence. minimal score correspond to the most probable sequences.
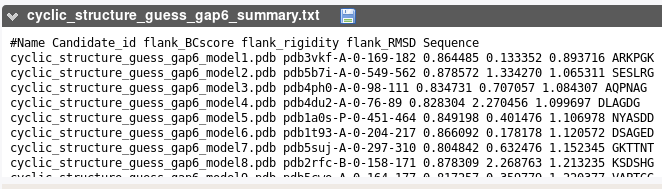
This section reports the sequence corresponding to the best candidates identified based on their geometry. For each, we indicate the PDB entry, the chain, and the residue numbers corresponding to the fragment, BCscore, RMSD, loop sequence.
This archive contains the 30 best predicted structures according to geometrical and sequence criteria are provided in PDB format. It is in the unix tar format compressed using gzip.
Once saved on your computer, enter for instance (unix) tar xzf AllModels.tgz to inflate the archive.
The result page of this demo can be accessed here.
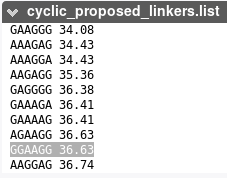
Example of the sequence logo generated for the un-cyclized peptide 1mxn. Experimentalists have reported a successful cyclization using a linker of 6 amino acids and of sequence GGAAGG, which is proposed by PEP-Cyclizer at rank 15 over 35.
 |
 |
The result page of this demo can be accessed here.
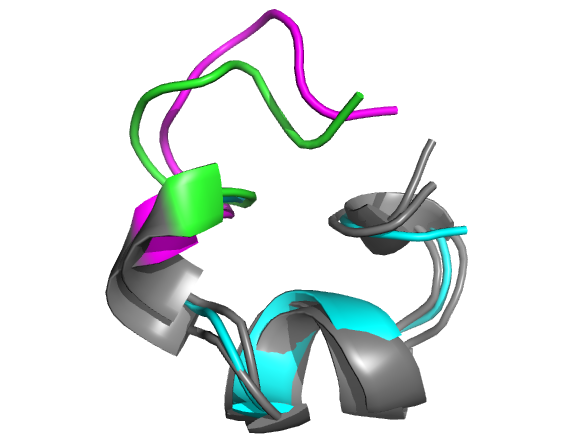
Example of the 3D modelling of the head-to-tail cyclic conformation of the alpha-conotoxin MII (PDB: 2ajw) starting from the un-cyclized structure (PDB: 1m2c) and a linker of sequence GGAAGG. Left: all models, as seen in the result page on the web. Right: Model 2 has an alpha carbon RMSD of 1.7 Angstroms (cyan: experimental un-cyclized conformation (PDB: 1m2c model 1); green: experimental conformation of the cyclic peptide (PDB: 2ajw); magenta: PEP-Cyclizer model 2; chain breaks correspond to the head-to-tail cyclization).
 |
 |
Peptide head-to-tail cyclization is assimilated to loop modeling where the loop consists of the linker between the C-terminus and the N-terminus of the un-cyclized peptide, considering flanks of 4 amino acids. The first linker residue corresponds to the residue next to the C-ter, etc.
PEP-Cyclizer is based on the identification of candidate fragments matching the geometrical conditions required for peptide head-to-tail cyclization. Such fragments are identified by mining the complete collection of protein structures of the PDB, using a protocol similar to that developped for protein loops [2], i.e. an approach searching for candidate fragments matching satisfactory geometrical contraints on the loop flanks [3][4]. Here, we search for fragments of size that of the linker, supplemented by flanks of 4 amino acids on both C-ter and N-ter directions (i.e. adding 8 amino acids), and we sort the candidates according to the RMSd calculated over the flanks only (8 amino acids - flank RMSD). We found that an acceptable RMSD cut-off is of 2 Angstroms. Candidates are sorted according to their flank RMSD, and a subset is accepted for further steps.
The candidate fragments are then filtered according constraints set by the user in terms of the linker sequence. Given explicitly the linker sequence, we select the fragments with a positive BLOSUM62 score with the linker sequence. Given constraints on amino acids types per position, since occurrences of sequences matching all constraints are unlikely, we select the fragments that have at least 50% of positions matching the constraints and having a positive BLOSUM62 score overall.
Given the candidates, 3D models of the cyclized peptide are built by superimposing the candidate linkers on the flanks, and repositioning the side chains using oscar-star [5]. Local geometry refinement is performed by minimizing the conformation of cyclized peptide using Gromacs [6].
The sequences of the accepted candidates are piled-up to generate a sequence logo. A Hidden Markov Model is fit on top of these sequences and we use the forward-backtrack algorithm to sample the sequences the most probable given the sequence constraints, and observed transitions between consecutive positions. Since both amino-acid frequencies and transitions are estimated from small ensembles, we make use pseudo-counts to un-bias them towards a uniform distribution.
Due to the weak number of cases where both the structures of un-cyclized and cyclized peptides are available, we have validated the approach on peptides of the cybase [7](66 entries with structure reduced to 36 after removing entries withou structures in the PDB, structures with gaps, etc), by mimicking the cyclization of un-cyclized peptides generated removing amino acids at N- and C-termini of the cyclic peptides. Considering the 10 (resp. the 30) best models, PEP-Cyclizer was able to return a model approximating the structure of the cyclized peptide at 1.84 (resp. 1.78) angstroms on average.
Extra validation could be be performed on a collection of 5 peptides for which the structures of both the un-cylized and cyclized peptides are available in the PDB, and 3 peptides for which the un-cylized conformation is available and information about successful linkers exist:
| un-cyclized | cyclized | linker size |
|---|---|---|
| 1m2c | 2ajw | 6 |
| 1m2c | 2ak0 | 7 |
| 2h8s | 4ttl | 6 |
| 2ew4 | 2j15 | 2 |
| 1ixt | 2mso | 3 |
| 1mxn | n/a | 4, 5, 6, 7 |
| 2jut | n/a | 2,3 |
| 2c9t | n/a | 3, 4, 5, 6, 7 (6 most stable) |
PEP-Cyclizer has been tested successfully using various OS / browser combinations, including:
[1] Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat, Weissig H, Shindyalov IN, Bourne PE.
The Protein Data Bank
Nucleic Acids Research, 2000; 28: 235-242.
[2]Karami Y, Guyon F, De Vries S, Tufféry P.
DaReUS-Loop: accurate loop modeling using fragments from remote or unrelated proteins.
Sci Rep. 2018; Sep 12;8(1):13673. doi: 10.1038/s41598-018-32079-w.
[3] Guyon F, Tufféry P.
Fast protein fragment similarity scoring using a Binet-Cauchy kernel.
Bioinformatics. 2014 Mar 15;30(6):784-91. doi: 10.1093/bioinformatics/btt618. Epub 2013 Oct 27.
[4] Guyon F, Martz F, Vavrusa M, Bécot J, Rey J, Tufféry P.
BCSearch: fast structural fragment mining over large collections of protein structures.
Nucleic Acids Res. 2015 Jul 1;43(W1):W378-82.
[5] Liang S, Zheng D, Zhang C, Standley DM.
Fast and accurate prediction of protein side-chain conformations.
Bioinformatics. 2011 Oct 15;27(20):2913-4.
[6] Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ.
GROMACS: fast, flexible, and free.
J Comput Chem. 2005 Dec;26(16):1701-18.
[7] Wang CK, Kaas Q, Chiche L, Craik DJ.
CyBase: a database of cyclic protein sequences and structures, with applications in protein discovery and engineering.
Nucleic Acids Res. 2008; Jan;36(Database issue):D206-10.