InterEvDock3
A docking server to predict the structure of protein-protein interactions using evolutionary information.
A docking server to predict the structure of protein-protein interactions using evolutionary information.
The InterEvDock3 service is integrated in the RPBS Mobyle Portal.
This website is free and there is no login requirement. Note that using the option for rescoring decoys using the Rosetta interface score is allowed only for non-commercial users (non-profit/academic/governement research groups). Commercial users are asked not to activate this option.
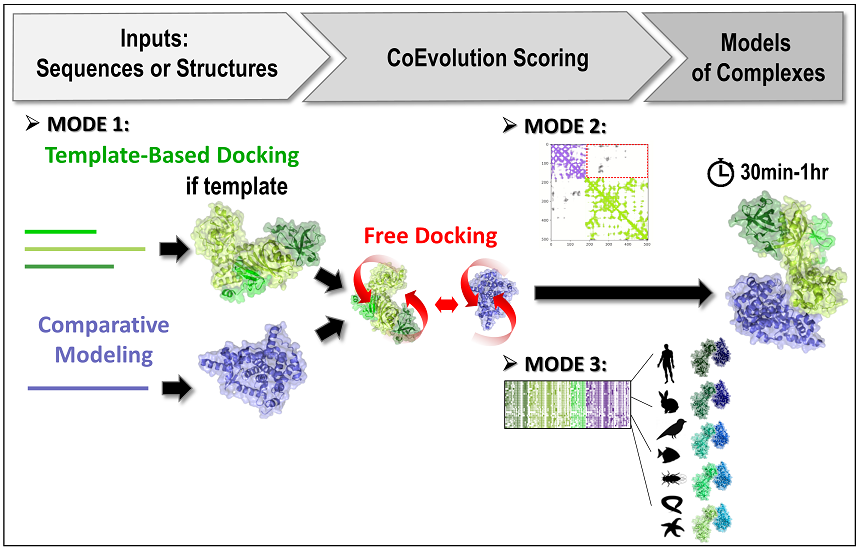
Two protein partners and their respective multiple sequence alignments are used to predict binding modes through a free docking procedure. Each partner can be a (set of) protein sequence(s) or a mono- or oligomeric structure. Comparative modeling will be performed if a (set of) sequence(s) is provided for one or both partner(s).
Watch the tutorial video to learn how to use InterEvDock3
The structural modeling of protein-protein interactions is key in understanding how cell machineries assemble and cross-talk with each other. When homologous sequences are available for both protein partners, it is very useful to rely on structures and multiple sequence alignments to identify binding interfaces. InterEvDock3 is a fully automated protein docking server that can start from input sequences or structures and take user constraints. If protein sequences are provided, template search and homology modeling (for monomers and assemblies) are performed on-the-fly. InterEvDock3 returns the most interesting docking models and suggestions of residues to target for mutagenesis studies.
InterEvDock3 runs a hybrid solution for template-based or free docking simulations under evolutionary constraints, thus ensuring the most efficient available approach is used at all steps of the structural prediction of protein assemblies. Joint alignments to exploit coevolution of the two binding partners are built by the server in a fully automated manner and integrated into model selection at an atomic detail through explicitly modeled homologs. In the InterEvDock3 server, the systematic docking search is performed using the FRODOCK2 programme [1] and the resulting models are re-scored with InterEvScore [2] together with the SOAP-PP atom-based statistical potential [3] and optionally also Rosetta's Interface Score [4]. The use of evolutionary information at atomic level was found to increase the confidence of free docking predictions [5].
InterEvDock3 is an update of InterEvDock [6] and InterEvDock2 [7]. Compared to its predecessors, InterEvDock3 features the following major upgrades:
When using this service, please cite:
InterEvDock3: A combined template-based and free docking server with increased performance through explicit modelling of complex homologs and integration of covariation-based contact maps. Submitted.Please also consider citing (for comparative modeling of complexes) [8] and/or (for free docking) [5].
If using free docking results, please also cite the FRODOCK programme which is used for the rigid-body docking step [1]. When using the results of SOAP-PP, please cite [3]. When using the results of Rosetta interface score, please cite [4]. When using the evolutionary conservation results obtained using Rate4Site (mapped onto all visualized models and written into the b-factor field of the PDB files provided for all models in the results zip archive) please cite [9].
When using comparative modeling results for complexes, please cite [8]. When using the automatic template search, please cite [10] and [11]. Since the comparative modeling protocol is based on OSCAR-star, please also cite [12].
InterEvDock3 supposes the interaction between the submitted proteins has been experimentally validated. It is not designed to predict neither the likelihood nor the strength of the interaction.
When free docking is performed, the two partners are modeled as rigid-body subunits. Selected models may have some clashes that can be released upon relaxation of the models.
In the free docking procedure, large conformational changes upon binding are generally not well predicted.
The simplest input consists in two fields to specify either the sequence(s) or the 3D structures of the partners to be docked. According to the input types, different pipelines will be activated in InterEvDock3 (cf. template-based modeling and free docking sections).
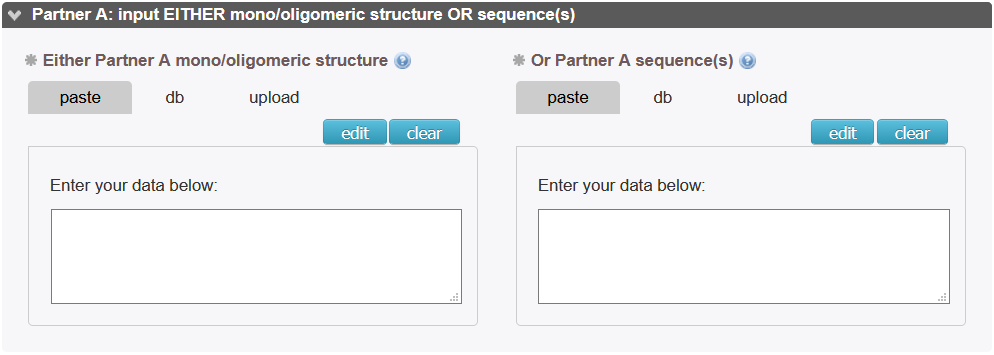
Partner A structure or sequence(s): Fill in one of the two fields.
It is possible to re-run docking using different conditions such as different templates (TBD MODE 1), constraints, different co-alignments (MODE 3) or different scoring options (including covariation constraints) (MODE 2). Specifying the identifier of a previous run (on the same two partners), the calculation will be much faster since the docking step will have already been calculated and only the scoring steps will need to be re-run. You can also use the hot restart feature to only minimize your output structures by setting the minimization field to "Yes" in the advanced options and keeping the exat same post-processing options as in the previous job.

If a sequence or a set of sequences is given as input for one or both partners, InterEvDock3 proceeds with its template-based modeling pipeline for the respective partners. The template is automatically chosen according to sequence identity, coverage and resolution of the potential template structures. At the end of the job, InterEvDock3 outputs a table of alternative templates that the user may want to try out. Templates are sorted by groups of same sequences that are covered (1st column) from the maximum to the minimum number of sequences (6th column). Each group has a representative template "t001" with the best id/coverage/resolution values (columns 4, 5 and 8 respectively). The 7th column specifies what sequences match which chains in the template as well as their individual pairwise sequence identities.
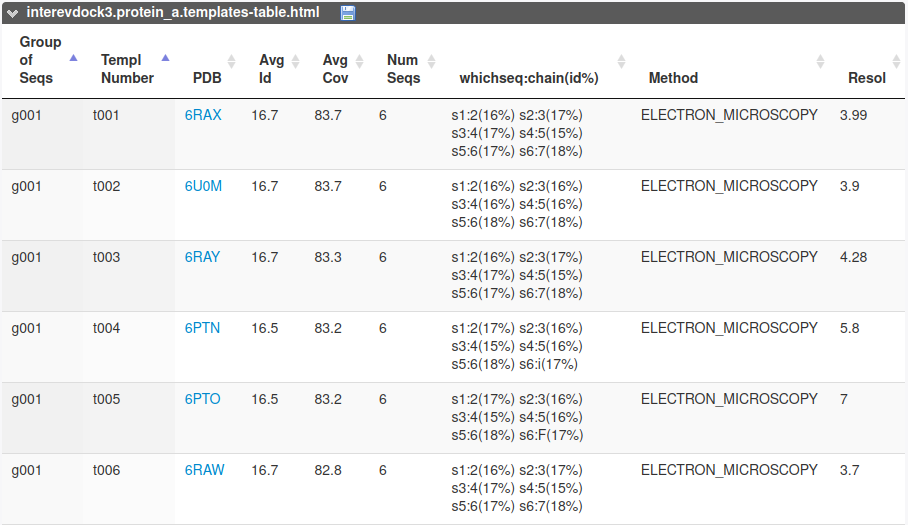
If the user wants to choose a different template from this list, he can do so by using the hot restart function of InterEvDock3 by specifying the previous job identifier and the new template's 4-letter code in the correspondings boxes.
This mode is advised only in case large multiple sequence co-alignments can be generated for the partners in interaction. If the user provides 2 input structures and a covariation map calculated with a program such as ComplexContact or trRosetta adapted to run on joint co-alignments, InterEvDock3 proceeds with free docking of the two partner structures guided by covaritation information found in the given contact maps. The format of the map and the different options available for the user to adjust are described here:
The user should input a contact_map as obtained for instance from a covariation-based method. First lines are the two sequences used to generate the covariation map in fasta format. Afterwards, each line is a coupling between residues as indexed in the sequences above with format
> sequence A XXXXXX > sequence B YYYYYY ires1 ires2 coupling_val ...
where ires1 and ires2 are the index of the residues in the fasta sequences and coupling_val is the confidence index as predicted in the covariation analysis methods.
Example:
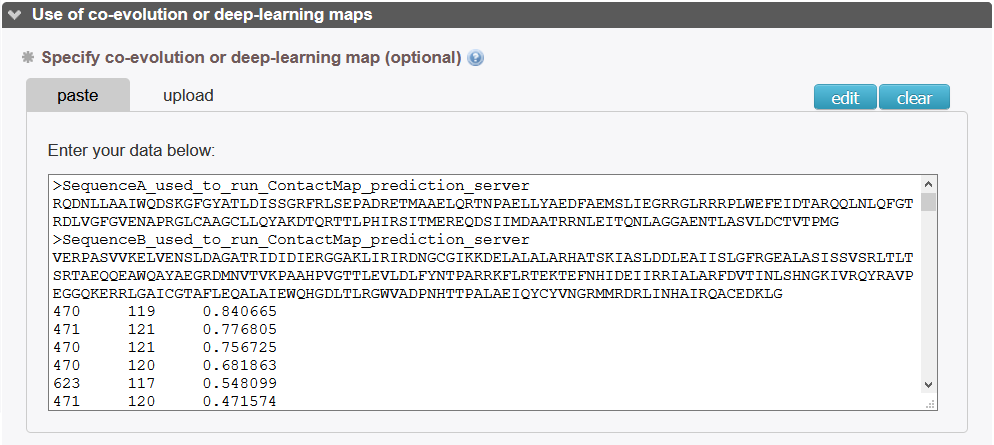
Of note, the server will align the fasta sequences with the pdb sequences. Users don't have to worry about the delimitations of their structures with respect to the sequences. Correspondance between the indexes will be automatically calculated.
The user can change the distance used to define if a contact is satisfied in the model. By default this value is set at 8A and corresponds to the distance between all heavy atoms in a pair of residues.
Contacts between residues close in sequence can be grouped together to prevent that too many redundant contacts bias the selection of the correct model. The size of the window used to group the contacts together is typically of 2, meaning two residues downstream and two upstream with respect to the resid1 and resid2 indicated in the map.
The user can change the number of decoys used to score the covariation-derived contact map. Generally, a first fast run with a small number of decoys can be tested (this typically take 30 min). If a more thorough sampling is required to increase the number of contacts likely to be taken into account in the models, the user can score all the decoys and the run might take several hours (typically about 3 hrs for large proteins).
At the end of the job, after downloading the results.zip archive, the user can open his results in PyMOL by double-clicking on the 'start_analysis_cmap.pml' script.
PyMOL will open and load the models as shown below.
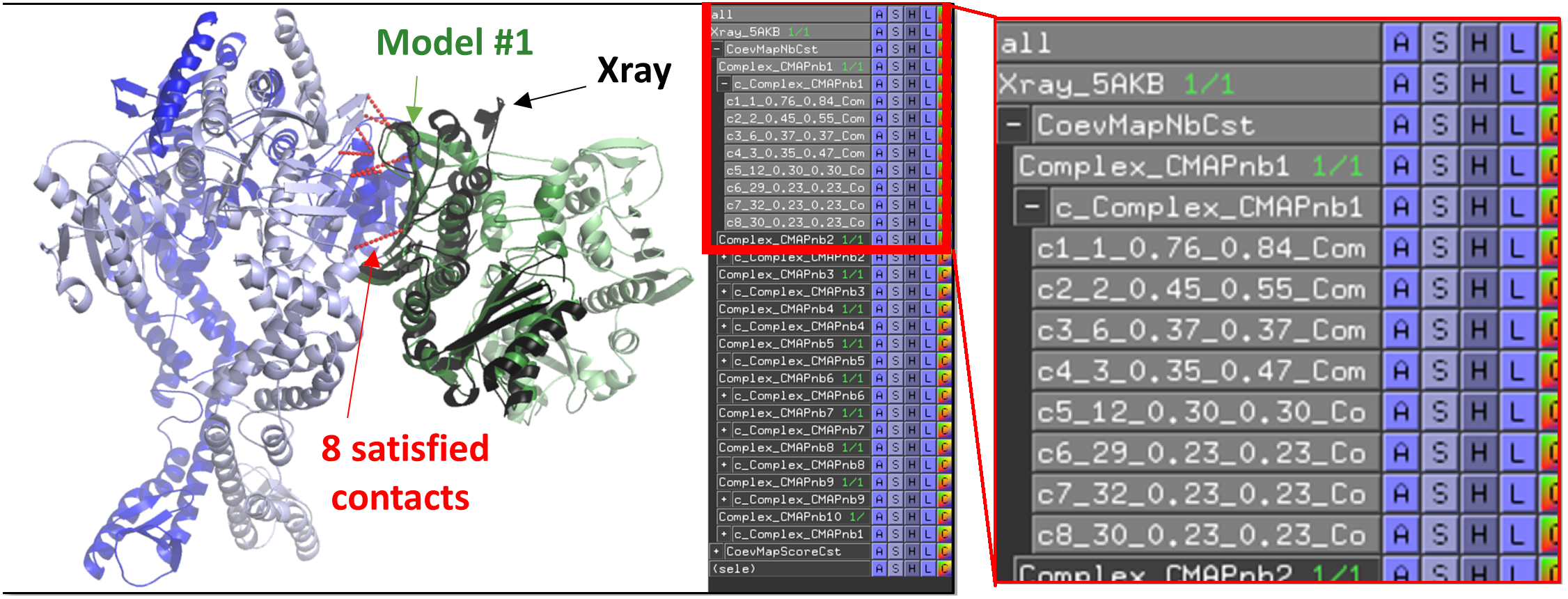
Satisfied contacts can be precisely analysed for every model and are indexed by their c1, c2, ..., cN names. Contact names have 5 elements separated with "_" symbols, corresponding to contact name, index of the line in the input contact map file, probability of the contact itself, probability of the reference contact in the group of redundancy which is used in the scoring, and model name (e.g. c1_1_0.76_0.84_Complex_CMAPnb1). Contacts can be displayed individually by toggling on and off their line in the panel.
This mode can be run even with shallow multiple sequence co-alignments generated for the partners in interaction (> 10 sequences). When two inputs are given (sequence or structure input for each partner), InterEvDock3 proceeds with free docking of the two partner structures. Co-multiple sequence alignments are automatically generated if not given. Several options are available to the user, the newest options are related to the newly implemented explicit interolog scoring described below. The user can also specify constraints to the docking if structures are given and use his own multiple sequence alignments.
A list of constraints can be optionally specified. Each constraint should contain three fields (filled or void) separated by colons (":"). The first field specifies a position on partner A, the second a position on partner B and the third the distance between both residues (contraint pair) or between the residue and any heavy atom on the opposite partner (single constraints). e.g.
11A:20B:7
if we want residue 11 on chain A in partner A to be at the interface as well as residue 20 on chain B in partner B with a maximum distance of 7 Å between the two. In order to specify a single constraint, you can just leave out the first or the second field (e.g. 11A::7 if you want residue 11 on chain A in Partner A at the interface with a maximum distance of 7 Å with any heavy atom on Partner B).
Positions are numbered according to the PDB numbering. Several constraints can be specified, separated by whitespace (space, tabulation or newline). A distance can be optionally specified for each constraint. The default distance is 8 Å for single residues and 11 Å for pairs. These constraints will be taken into account in the docking process to keep only models having this position or pair of positions at the complex interface. Constraints will be checked prior to applying and constraints involving residues not present or buried in the structural model will be excluded. Note that when several constraints are provided, they are considered cumulative ("AND" not "OR") i.e. docking models will be filtered to retain only solutions that verify all constraints.
In order to avoid round-trips outside the browser, it is possible to use the on-line NGL Viewer viewer to quickly identify residues.
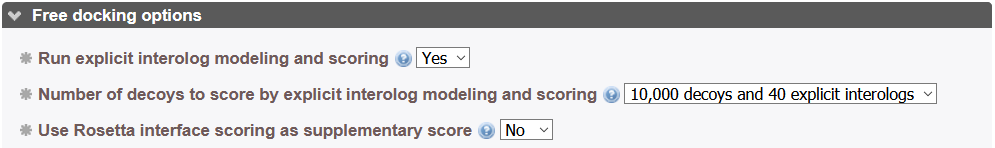
If the miinimization option is set to "Yes", structures at the end of a docking round are minimized using the Gromacs_py library[14]. Please take into account that minimization can take 10 to 20 minutes or more according to protein size. We recommend that you run minimization in a second step using the hot restart feature of InterEvDock3 to minimize all models of a previous job if satisfied with the results.
InterEvDock3 runs last from 15 minutes to over 1 hour depending on the size of the input partners and the server load. The user will be able to follow the progression of his job thanks to the Progress report field of the job page. Errors related to the input data specified are also reported in this field.

When the job is finished, an interactive page will allow the user to browse the best generated complexes (see below Visualization and post-processing). A zip archive is also provided containing the PDB files of the predicted models and any additional information such as individual and consensus scoring information, consensus interface residue probabilities, multiple sequence alignments used in scoring or detailed template lists when relevant and also contains pymol visualization scripts for local analyses.
The best models can be explored using the NGL applet.
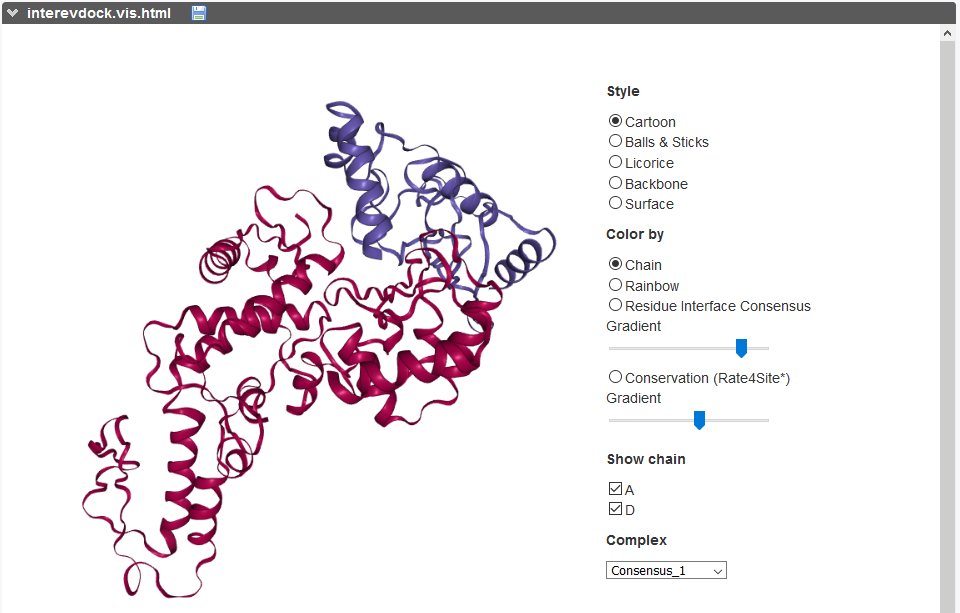
The predicted interface residues outputted after the free docking protocol can also be visualized on both proteins as a color gradient (from green to white for high to low probability to be at the interface).
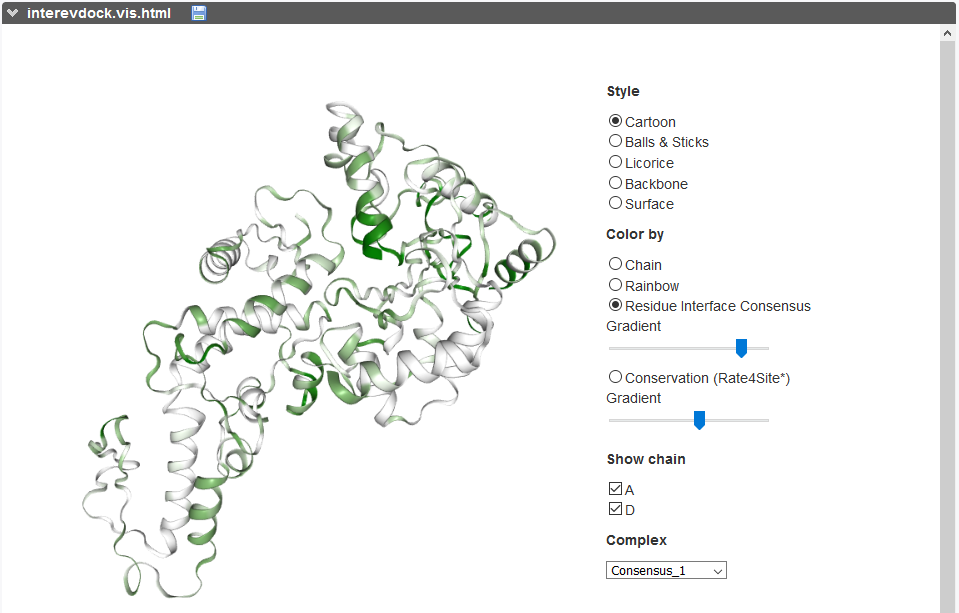
The evolutionary conservation of each partner (calculated with Rate4Site [9] in the free docking protocol) can be visualized on both partners as a color gradient, from red (more conserved) to white (more diverse) through beige (mild conservation).
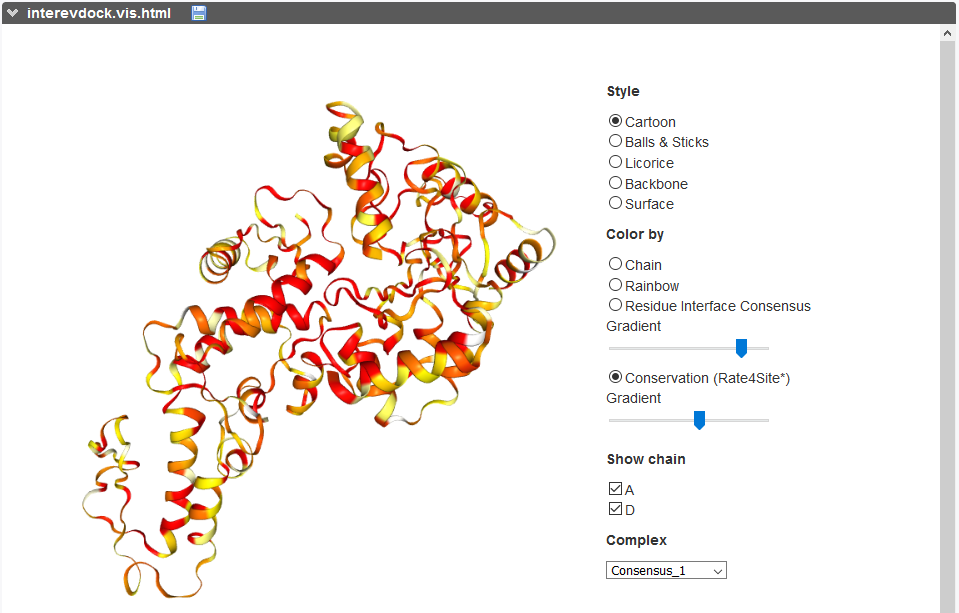
A PyMOL script provided in the results zip archive for each free docking run automatically loads the 30-50 models (according to selected options) from the results zip file and colors them by interface residue consensus and evolutionary conservation.
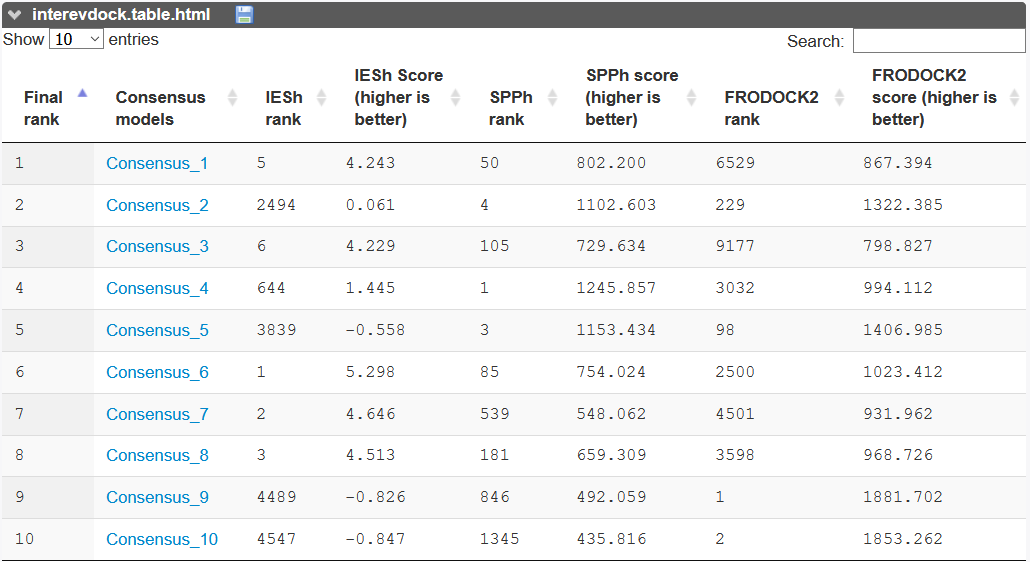
In the free docking mode, all calculated scores are reported in a table for the 10 best complexes.

If constraints were specified in input, information about each constraint satisfaction is presented as a table.
Accessible from InterEvDock3 page by selecting an option from the Demonstration Mode dropdown menu. By setting this option, InterEvDock3 will load pre-configured test cases.
There are three demonstration mode cases:
Details on the performance of these demonstration examples are provided below.
The result page of this demo can be accessed here.
Starting from input sequences only given in the Partner A input box, InterEvDock3 builds a model of the Lachancea thermotolerans SHU1-SHU2 complex through a template-based docking procedure. The automatic template search found the Saccharomyces cerevisiae SHU1-SHU2 interolog (5XYN) with 28% and 36% sequence identity and a total coverage of 98.3% with L. thermotolerans. The resulting output model has a 1.86 Å RMSD ("Medium" quality according to the CAPRI criteria) with its template structure.
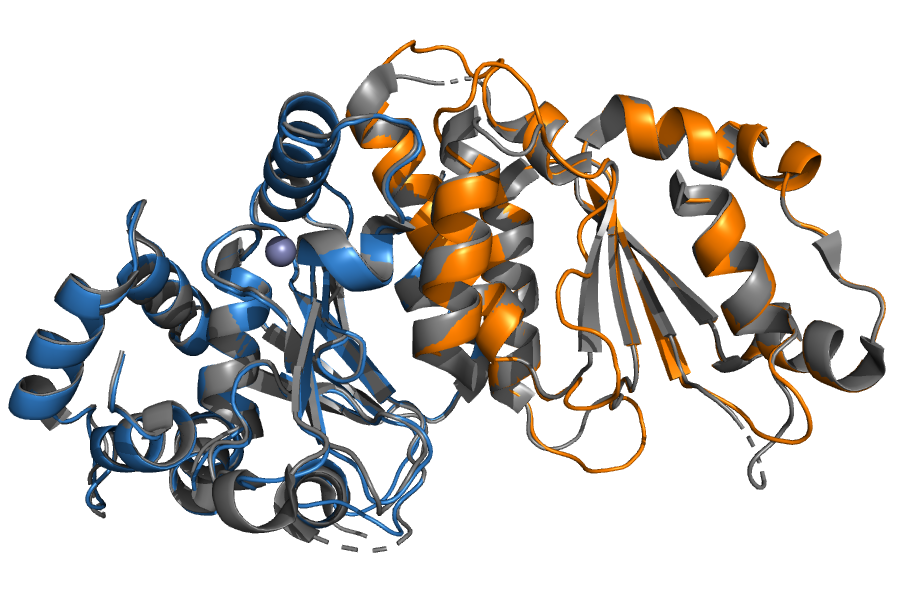
Example of template-based modeling by InterEvDock3. Output of L. thermotolerans SHU1-SHU2 complex (in orange and blue cartoon) modeled using an interolog structure from S. cerevisiae (5XYN chains C and D in gray cartoon).
The result page of this demo can be accessed here.
InterEvDock3 uses free docking between two binding partners to search for the best assemblies matching the interfacial contacts as predicted by covariation-based methods such as Complex-Contact or CCMpred. These methods generate contact maps based on the predicted evolutionary couplings but do not embed any docking method to generate compatible structures. The trRosetta program can also be used to generate these maps by adapting the trRosetta package to run on two joint co-alignments. Here, we illustrate how the interaction between MutS homodimer and MutL homodimer from Escherichia coli can be predicted using their structures as input and the predicted interfacial contacts obtained using the predictions from Complex-Contact which combines co-evolution and deep learning techniques.
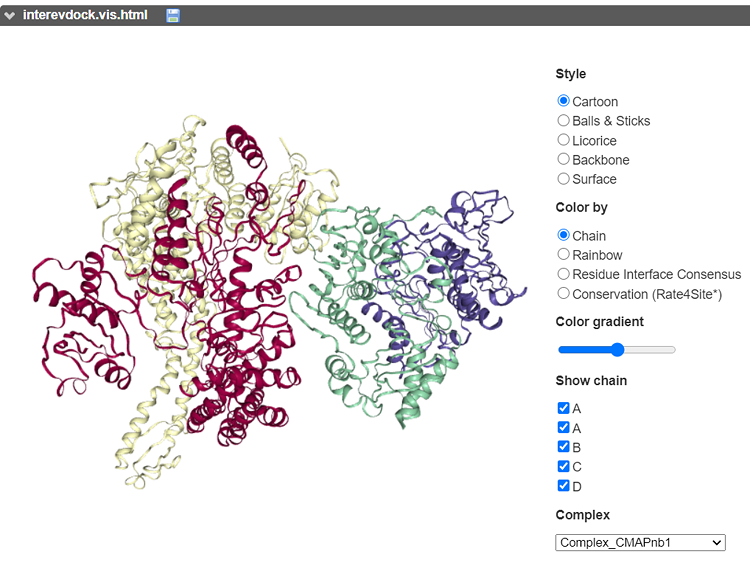
Example of free docking exploiting the contact map predicted by co-evolution and deep learning techniques (using Complex-Contact). Output of E. coli MutS (red/yellow) and MutL (blue/green) homodimers free docking as displayed in the server. The presented model was ranked #1 ("Medium" according to the CAPRI criteria).
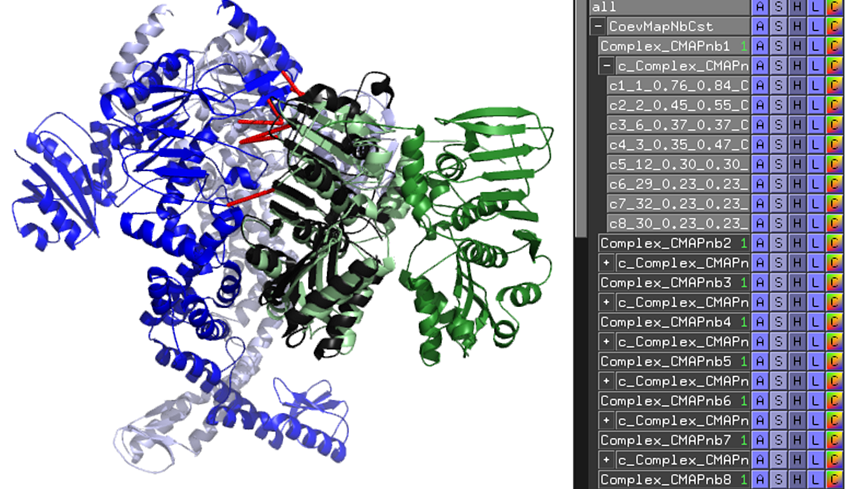
A PyMOL script "start_analysis_cmap.pml" can be downloaded from the results.zip. Double clicking on the script opens the output representing E. coli MutS and MutL free docking models. The contacts respected in the contact map are represented as red sticks (see below for interpretation details). Here, the best model #1 is compared to the reference crystal structure (5AKB in gray cartoon).
On the PPI4DOCK benchmark, our updated approach using evolutionary information at the atomic level provides a general performance boost (see the Benchmark section). The new scoring scheme is based on a 3-way consensus using atomic-level homology scoring. Here, we illustrate this with with two input structures and associated joint multiple sequence alignments for case 1c4z_AD from the PPI4DOCK benchmark.
The result page of this demo can be accessed here. Results using the same input but turning OFF explicit atom-based evolutionary scoring can be accessed here.
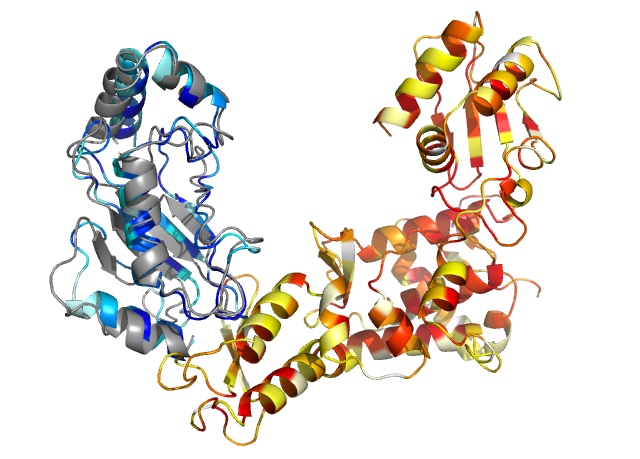
Best model returned by InterEvDock3. Color darkness reflects conservation of the model in shades of yellow and red for chain A and blue for chain D. This model was returned as top 3 by the consensus and top 6 by atomic-level InterEvScore (IESh) and has an iRMSD of 1.51 Å and DockQ of 0.71 ("Medium" quality according to the CAPRI criteria) with the reference crystal structure (1c4z in gray).
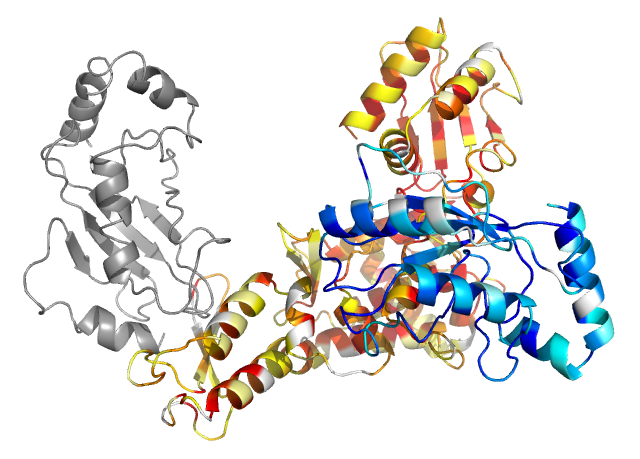
Best model returned in the top 10 consensus by InterEvDock3 when not using atomic-level homology integrated into scoring (similar to InterEvDock2). This model was returned as top 3 by the consensus and top 4 by SOAP-PP and has an iRMSD of 16.78 Å and DockQ of 0.04 ("Incorrect" according to the CAPRI criteria) with the reference.
| Name | Description |
|---|---|
| 6-subunit human inner kinetochore | A challenging low-identity template-based docking example. InterEvDock3 managed to predict a complex with all six subunits despite sequence identities between human and yeast ranging from 9 to 13% for all subunits. |
| ComM helicase competence protein in H. pylori | An example to illustrate the combination between template-based modeling and free docking guided by a covariation-derived contact map. ComM is composed of two domains belonging to well-known superfamilies but for which a full-length structural template including both domains is lacking. With InterEvDock3, we were able to model the hexameric assemblies for each domain separately (~20% sequence identity) and propose a decent full structural model thanks to free docking of these models guided by the evolutionary signal contained in contact maps calculated by the trRosetta server. |
| SYCE2-TEX12 (T163 in CAPRI) | CAPRI target T163 is a complex between 2 homodimers. SYCE2 homodimer could be modeled with template-based docking in InterEvDock3 using 6H3A (a template structures that was available at the time of the CAPRI challenge). The model was docked against a single helix extracted from TEX12 homodimer structure (PDB:6HK9) and guided by covariation information calculated with RaptorX. The best model satisfies the largest number of inter-molecular contacts between SYCE2 and TEX12 taking into account the ambiguities due to symmetrical arrangement of the SYCE anti-parallel dimer. Another run using as input of the free docking, the structures of SYCE2 and TEX12 subunits as crystallized in the complex 6R17 can also be analyzed. With this docking using bound subunits, 56% of the contacts predicted amond the Top50 most probable contacts were found respected in #1 model while there were 41% of them in the #1 model of the docking using modeled subunits as described above. |
| 6-subunit COMPASS complex in S. pombe | Template-based docking of the COMPASS complex based on K. lactis interolog (6BX3) with some subunits sharing less than 20% sequence identity. |
InterEvDock3 takes as input either the structures of two protein partners to be docked: experimental or modeled structures, possibly multimeric, or sets of sequences. If a single set of sequences is provided as Partner A, then comparative modeling will be performed to build a structural model for this set of sequences.
Given a (set of) sequence(s), for a given partner, the server runs several steps to generate a structural model:
Given the structures (either input by the user or modeled from input sequences), the server runs several steps to propose a selection of 10 most likely models for each score as well as 10 consensus models and 5 most likely interface residues on each protein:
If covariation-based pairwise constraints are provided by the user (e.g. derived from deep-learning-assisted predictors such as ComplexContact), the scoring process is bypassed and replaced by the counting of the predicted contacts satisfied in every docking model. The server runs several steps to match the residue indexes between the contact map (derived from sequence) and the input structures, then counts contacts using several options that are described below:
Homology is cunningly derived to the atomic-level thanks to a basic and very conservative comparative modeling of homologous sequence pairs in the provided or generated coMSAs using OSCAR-star [12]. This new representation of evolutionary information is directly compatible with scores such as SOAP-PP and Rosetta's Interface Score and proved to significantly improve sucess rates of individual scores and consensuses of these scores [5].
Performance of InterEvDock3 was assessed on 812 complexes from the PPI4DOCK database [16] for which the structures of the free proteins (unbound) can be modeled and for which evolutionary information could be retrieved [2]. A table of all 812 benchmark cases is available here.
The InterEvDock3 server predicts an "Acceptable" or better solution in the consensus top10 for 268 out of 812 test cases in default mode (IED3-atom default, 33%) and for 284 out of 812 in slow mode (IED3-atom slow, 35%). As expected, this top10 success rate decreases with increasing difficulty of the docking cases: 45.4% for very easy targets, 34.5% for easy targets, 12.7% for hard targets and 9.1% for very hard targets according to PPI4DOCK difficulty classification (51.7, 36.1%, 11% and 4.5% respectively).
The InterEvDock3 server also predicts residues making contacts at the interface of a complex based on the analysis of all the interfaces of the top 10 decoys for all scores composing the consensus (30-50 models depending on the options). In 90.8 % of the 812 test cases, at least one residue out of 10 was correctly predicted as present at the interface, providing very useful hints to guide mutagenesis experiments to disrupt a complex of interest. Of note, there is little decrease in precision from the rigid-body to the difficult cases (success rate is 92.5% for very easy targets, 91.8% for easy targets, 84.7% for hard targets and 86.4% for very hard targets according to PPI4DOCK difficulty classification). Predictions of the InterEvDock3 server can thus also be used as a prior to constrain more thorough docking simulations requiring flexibility in order to model the correct orientation between two binding partners. In that perspective, in 53.4% of the cases, at least one correct residue is predicted on both sides of the interface (61.5% for very easy targets, 54.6% for easy targets, 37.3% for hard targets and 50.0% for very hard targets according to PPI4DOCK difficulty classification). When considering only the top 1 predicted residue on each chain, at least one of the two predicted residues is correct in 77.5% of the cases and both are correct in 37.1% of the cases, highlighting the practical value of InterEvDock3 residue prediction. All those results are significantly higher than a reference interval given by random selection of residues on the surface of the protein.
FRODOCK 2.0: fast protein-protein docking server.
Bioinformatics. 2016; 32(15):2386-8.
InterEvScore: a novel coarse-grained interface scoring function using a multi-body statistical potential coupled to evolution.
Bioinformatics 2013; 29 (14):1742–1749.
Optimized atomic statistical potentials: assessment of protein interfaces and loops.
Bioinformatics. 2013; 29(24):3158-66.
Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations.
J Mol Biol. 2003;331:281-99.
Atomic-level evolutionary information improves protein-protein interface scoring.
bioRxiv 2020.10.26.355073.
InterEvDock: A docking server to predict the structure of protein-protein interactions using evolutionary information.
Nucleic Acids Res. 2016 Jul 8;44(W1):W542-9.
InterEvDock2: an expanded server for protein docking using evolutionary and biological information from homology models and multimeric inputs.
Nucleic Acids Res. 2018 Jul 2;46(W1):W408-16.
Probing Protein Interaction Networks by Combining MS-Based Proteomics and Structural Data Integration.
J. Proteome Res. 2020 Apr 27;19(7):2807-2820.
Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues.
Bioinformatics. 2002; 18 Suppl 1:S71-77.
HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment.
Nat Methods. 2011;9(2):173-5.
Protein homology detection by HMM-HMM comparison.
Bioinformatics. 2005; 21(7):951-60.
Fast and accurate prediction of protein side-chain conformations.
Bioinformatics. 2011 Oct 15;27(20):2913-4.
DaReUS-Loop: a web server to model multiple loops in homology models.
Nucleic Acids Res. 2019 Jul 2;47(W1):W423-8.
Gromacs_py
Github repository
BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences.
FEMS Microbiol Lett. 1999;174(2):247-50.
PPI4DOCK: large scale assessment of the use of homology models in free docking over more than 1000 realistic targets.
Bioinformatics. 2016; 32(24):3760-3767.
This work was supported by the French Infrastructure for Integrated Structural Biology (FRISBI) [ANR-10-INSB-05-01], the Agence Nationale de la Recherche through grants CHIPSET [ANR-15-CE11-0008-01] and ESPRINet [ANR-18-CE45-0005-01], the French Institute for Bioinformatics (IFB) [ANR-14-2011-IFB], the IdEx Université de Paris [ANR-18-IDEX-0001], the IDEX Paris-Saclay [IDI 2017], the MINECO [BFU2016-76220-P], the AEI/FEDER and UE [PID2019-109041GB-C21].