pepATTRACT
Blind, proteome-wide peptide-protein docking.
Blind, proteome-wide peptide-protein docking.
Peptide-protein interactions are ubiquitous in the cell and form an important part of the interactome. Computational docking methods can complement experimental characterization of these complexes, but current protocols are not applicable on the proteome scale.
We have previously introduced pepATTRACT [1], a novel docking protocol that is fully blind, i.e. it does not require any information about the binding site. Nevertheless, its performance is similar or better than state-of-the-art local docking protocols that do require binding site information.
Here we present a Web server for pepATTRACT, carrying out only the rigid-body stage of the protocol, performing docking runs in about 10 minutes. Combined with the fact that it is fully blind, this makes the Web server well-suited for proteome-wide in silico protein-peptide docking experiments.
Access the service through the RPBS Mobyle portal:
When using this service, please cite the following reference:
Schindler CEM, De Vries SJ, Zacharias M.
Fully blind peptide-protein docking with pepATTRACT.
Structure. 2015 Aug 4;23(8):1507-15.
De Vries SJ, Rey J, Schindler CEM, Zacharias M, Tuffery P.
The pepATTRACT web server for blind, large-scale peptide-protein docking.
Nucleic Acids Res. 2017 Apr 29.
The Web server requires two inputs:
For more information about the file formats, please read the Mobyle Tutorials section: here for 3D formats and here for sequence format.

This section will incrementally provide information about job progression and errors if any. A typical run should produce a report similar to the following :

Errors related to the input data specified are also reported in this field.
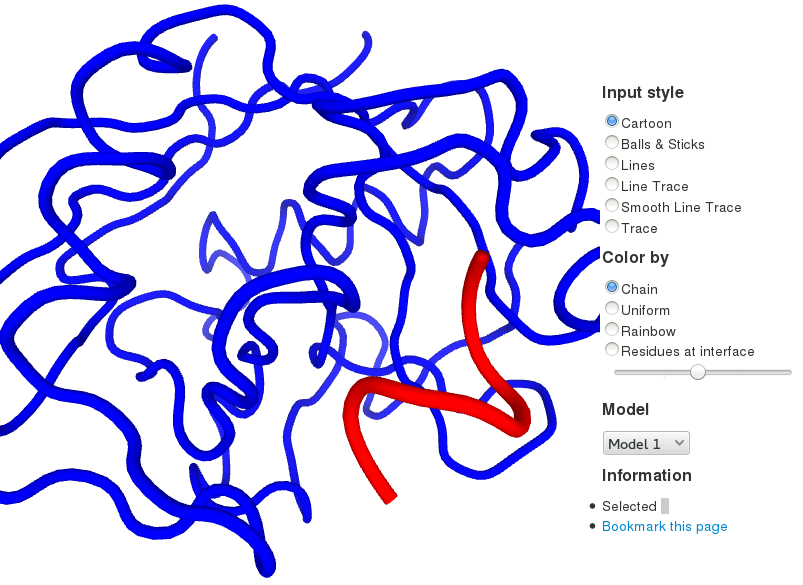
The server returns 50 models, with each model being the lowest-energy structure of a docking cluster. These models can be downloaded (click the floppy disk icon in the result.pdb section). They are also visualized directly in the browser (in the Visualization section). The interactive display relies on the PV - JavaScript Protein Viewer
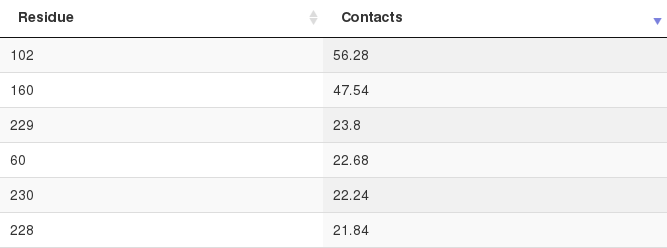
The interface propensity of every residue is also given (also see the Interface prediction performance below):
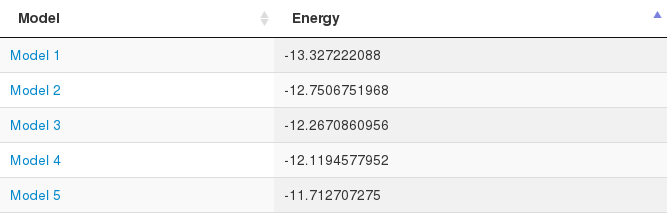
Finally, the ATTRACT force field energy of each model is shown in a table. Note that this energy should be used only to identify the correct model, it cannot be used to predict binding affinities.
The performance of the pepATTRACT Web server was tested on the 31 complexes from the peptiDB benchmark [2] where unbound receptor structures are available and where the stoichiometry of the biological unit is 1:1. For 13/31 complexes, at least one of the 50 models had an interface RMSD (iRMSD) of better than 2 Å. Among the top 10 models, this was achieved for 11 complexes. The full pepATTRACT protocol achieves this for 16 cases.
Despite the reduced pepATTRACT Web server protocol does have a somewhat decreased performance compared to the full pepATTRACT protocol, which includes a flexible refinement using iATTRACT [1] and a molecular dynamics refinement and re-scoring, it does not imply irrelevant poses as illustrated in the Interface prediction performance section below. Thus, in its present implementation, the pepATTRACT Web server is aimed towards for rapid, large-scale in silico docking experiments. In contrast, the computationally intensive full pepATTRACT protocol is meant to be run locally, and can be configured via the existing Web interface at http://www.attract.ph.tum.de/services/ATTRACT/peptide.html. The possibility to extend the pepATTRACT Web server to include the iATTRACT step is however questioned.
The Web server also returns an analysis of the protein residues that are most prevalent in protein-peptide contacts among the top 50 docking models (before clustering). The interface propensity (the average number of protein-peptide contacts) of each residue is visualized in a table.
In the docking model visualization, the interface propensity can be projected onto the receptor protein (“Residues at interface” coloring option). When the top N residues with the highest interface propensity are selected (with N being the total number of interface residues), both the sensitivity and the specificity of the interface prediction are of close to 46%.
The adaptor protein Hop mediates the association of the molecular chaperones Hsp70 and Hsp90. The TPR1 domain of Hop specifically recognizes the C-terminal heptapeptide of Hsp70, ending with the motif EEVD. The crystal structure of the TPR1-peptide complex (PDB code: 1ELW) shows the peptide in an extended conformation, spanning a groove in the TPR domain. The hydrophobic contacts with the peptide are critical for specificity. Starting from the conformation of the unbound protein (PDB: 1A17), pepATTRACT identifies the correct binding site with high confidence (left), and the best pose #13 has an interface RMSD of 0.83 Å towards the experimental peptide pose (right).
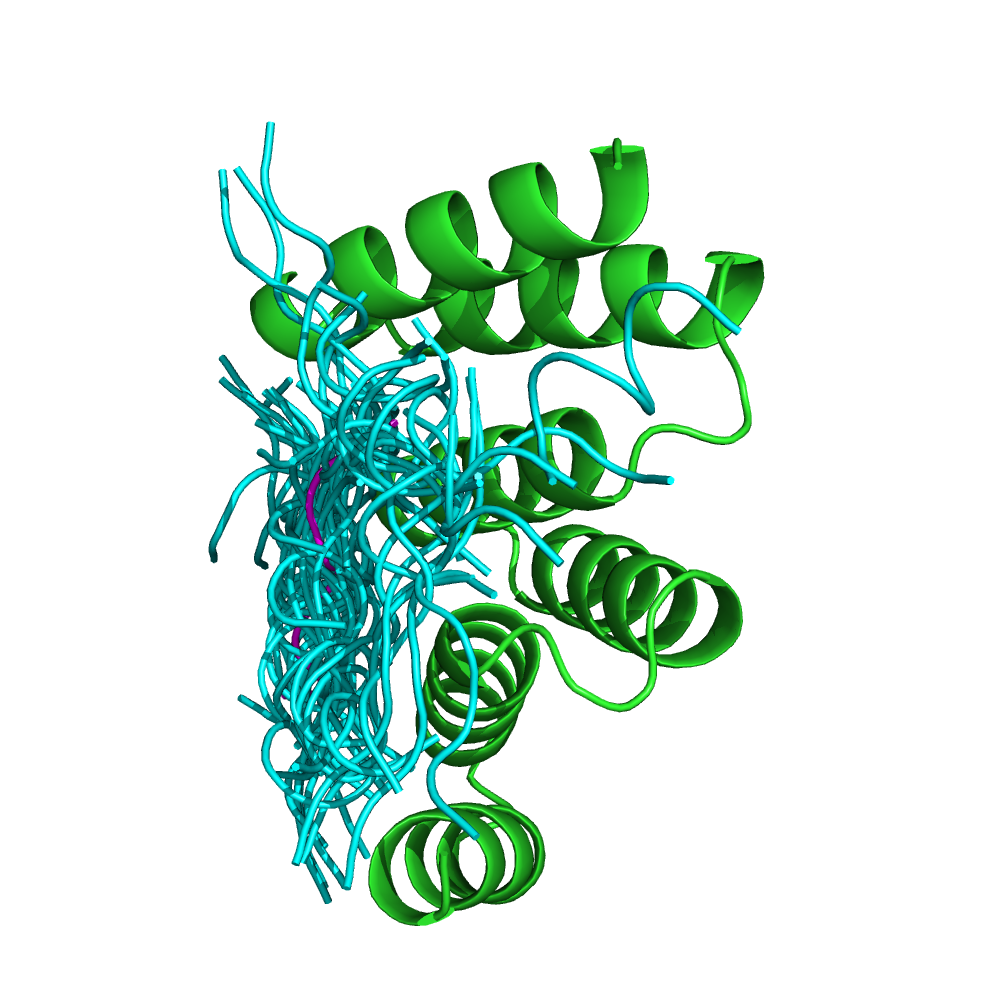
Experimental complex structure of the unbound conformation of the receptor (1A17) with the 50 peptide best poses. green: protein; cyan: peptide. magenta: experimental peptide conformation.
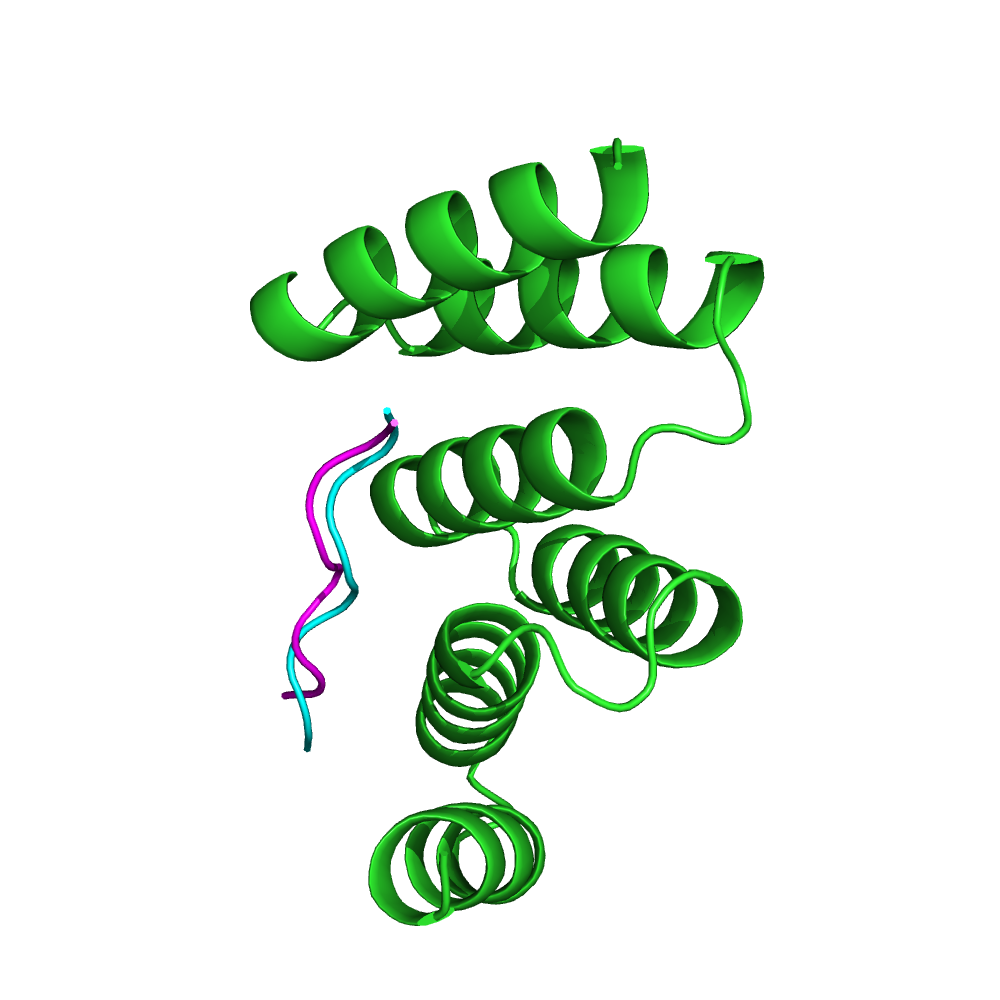
Peptide pose 13 (iRMSD 0.83 Å). magenta: experimental peptide pose.
GGA proteins are critical for the transport of soluble proteins from the trans-Golgi network (TGN) to endosomes/lysosomes by means of interactions with TGN-sorting receptors. The GGA VHS domain recognizes these receptors via acidic-cluster dileucine (ACLL) sequences. The GGA1 VHS domain has been crystallized in complex with the ACLL sequence of the mannose 6-phosphate receptor (PDB: 1JWG). Starting from the conformation of the unbound protein domain (PDB: 1MHQ), pepATTRACT favors the correct binding site (left). The top-ranked pose has an interface RMSD of 1.490 Å towards the experimental peptide pose, and the best pose #47 has an interface RMSD of 0.910 Å (right).
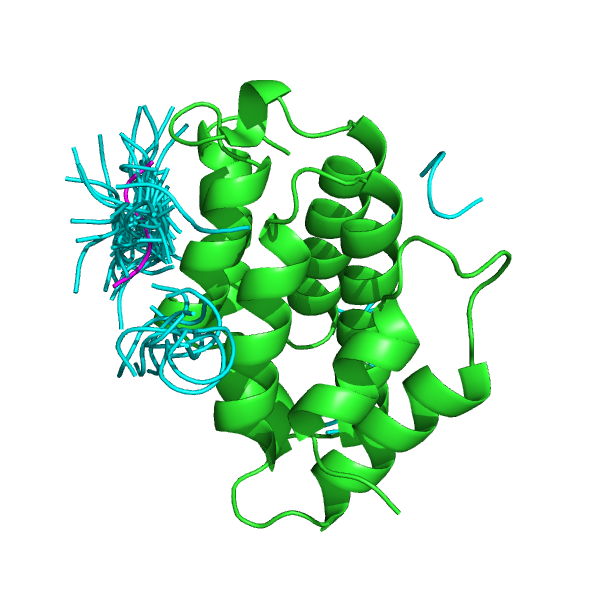
Experimental complex structure of the unbound conformation of the GGA VHS domain (1MHQ) with the 50 peptide best poses. green: protein; cyan: peptide.
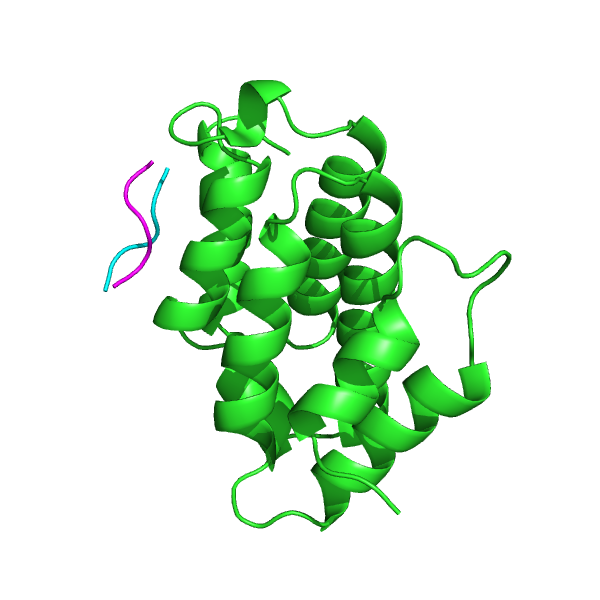
Peptide pose 47 (iRMSD 0.91 Å). magenta: experimental peptide pose.
[1] Schindler CEM, De Vries SJ, Zacharias M.
Fully blind peptide-protein docking with pepATTRACT.
Structure. 2015 Aug 4;23(8):1507-15.
[2] London M, Movshovitz-Attias D, Schueler-Furman O.
The structural basis of peptide-protein binding strategies.
Structure. 2010 Feb 10;18(2):188-99.
[3] De Vries SJ, Rey J, Schindler CEM, Zacharias M, Tuffery P.
The pepATTRACT Web server for blind, proteome-wide peptide-protein docking.
Nucleic acids research 45 (W1), W361-W364