ProPHet
Probing Protein Heterogeneity.
Probing Protein Heterogeneity.
The ProPHet [1,2] program combines a coarse-grain [3] / elastic network (ENM) protein model and a Brownian Dynamics algorithm to compute protein local rigidity on the residue level.
Access the service through the RPBS Mobyle portal:

Three stages of coarse graining citrate synthase, with the final ENM representation in panel (c) (picture from ref. [2]).
The program uses a PDB structural file as a starting point and will produce a rigidity profile of the protein under study with a force constant value for each residue in the protein.
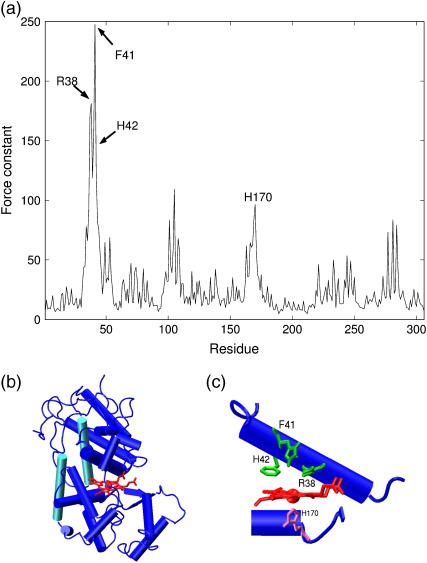
(a) The force constant profile of horseradish peroxydase. (b)(c) A close-up view of the heme binding site with the rigid catalytic and iron binding residues in green and pink (picture from ref. [4]).
As an input you will have to upload a pdb file of the protein structure you want to study. If this is a file from the RSCB PDB, make sure it contains only one monomer of your protein, since the program reads the complete pdb file and will consider that every atom belongs to the same unique protein chain. Otherwise, you will have to specify which protein chain you want the program to treat.
Other options are the following:
There are different ways to use the rigidity profiles produced by ProPHet calculations:
For further details about ProPHet you can also see refs. [1,2,4]
[1] Lavery R, Sacquin-Mora S.
Protein mechanics: a route from structure to function.
J Biosci. 2007 Aug;32(5):891-8.
[2] Sacquin-Mora S.
Motions and mechanics: investigating conformational transitions in multi-domain proteins with coarse-grain simulations.
Mol. Simul. 2014, 229-236.
[3] Zacharias M.
Protein-protein docking with a reduced protein model accounting for side-chain flexibility.
Protein Sci. 2003 Jun;12(6):1271-82.
[4] Sacquin-Mora S, Lavery R.
Investigating the local flexibility of functional residues in hemoproteins.
Biophys J. 2006 Apr 15;90(8):2706-17. Epub 2006 Jan 20.
[5] Sacquin-Mora S, Laforet E, Lavery R.
Locating the active sites of enzymes using mechanical properties.
Proteins. 2007 May 1;67(2):350-9.
[6] Sacquin-Mora S.
Bridging enzymatic structure function va mechanics: a coarse-grain approach.
Methods Enzymol. 2016;578:227-48.
[7] Sacquin-Mora S.
Fold and flexibility: what can protein's mechanical properties tell us about their folding nucleus ?
J R Soc Interface. 2015 Nov 6;12(112).
[8] Bocahut A, Bernad S, Sebban P, Sacquin-Mora S.
Frontier residues lining globin internal cavities present specific mechanical properties.
J Am Chem Soc. 2011 Jun 8;133(22):8753-61.
[9] Oteri F, Baaden M, Lojou E, Sacquin-Mora S.
Multiscale simulations give insight into the hydrogen in and out pathways of [NiFe]-hydrogenases from Aquifex aeolicus and Desulfovibrio fructosovorans.
J Phys Chem B. 2014 Dec 4;118(48):13800-11.