the Protein Picture
Generator
This service is
aimed at providing an easy way to generate pictures of protein
structures, with the concern of integrating the most frequently used
concepts of the molecular graphics fields. The design of the interface
results from discussions with potential users about the features they
would like to be implemented. The underlying machinery acts like a
black box, but some understanding about the features of the interface
might come from the knowledge that we presently use the Dino program to produce the
images, coupled with external programs such as stride
(secondary
structure determination), hbplus
(hydrogen bond determination) or msms
(molecular surface
calculation).
History
- June-August 2004: First implementation by Cedric Binsiti and Salim
Ahmed.
- September 2004: First development release of the service,
restrained access. Many improvements, in particular, users express the
need for animations.
- Late October 2004: Second development release. Access set free to
world, information to some Paris teams.
- December 2004: Pre-production release. Minor bug fixes. Server can
treat several simultaneous requests.
- January 2005: Full production release, version 1.0.
- March 2005: Bug fixes (DNA), orientation previewer based on JMol, version 1.1.
- December 2005: Version 1.2, new previewer (sliders to reach interactive pre-orientation)
- June 2006: Version 1.21, small bug fix for side chains management in the default representation.
1. Design of the form
2. Data specification
3. Default representation
4. Supplementary representations
5. Process
6. Advanced parameters
7. Citations
8. Feedback
9. Gallery / examples
1.
Design of the form:
The
form is divided in four sections concerning respectively:
- Advanced parameters allowing to customize
most
of the drawing
parameters such as the color patterns, etc, can be accessed at the end
of the form.
2.
Data
specification:
The data to render
must
be in the Protein Data Bank format
(PDB). It can be specified either on the form
or
a PDB Id , or by
uploading the data. PDB Ids must be on the form: 1tim for
the whole 1tim PDB entry, or 1timA for the chain A of the 1 tim entry. PDB identifier (the first 4 characters)
are not case sensitive, hence 1Tim, 1TIM or
1tim are equivalent.
Uploading data or specifying a PDBId,
you can also specify a list of chain Ids
in the "Chains to visualize"
field (e.g. AB, or A,B to select chains A and B of the PDB entry).
Specifying "All" or nothing will result in keeping all chains. Blank is a valid chain
identifier. Chain identifier are case sensitive.
1timA and 1tima are NOT equivalent.
Currently,
it is possible
to render only one PDB file. We plan to improve this soon. To make an image including several PDB
entries, you can however concatenate them into one file, labeling
each file using
separate chain Ids.
3. Default
representation:
3.1
Title:
This character string eventually specified here will
be automatically inserted into the picture. Multiple line text are
possible using "\n" line break convention. Advanced parameters allow
the customization of its aspect (font family, size and face) and
position.
3.2
Display:
This is to specify what
will be displayed from the whole molecule.
- backbone:
specify if you want or not to
represent the backbone (should also work soon for nucleic acids).
- side-chains:
specify if you want or not to represent
the side chains (should also correspond soon to the bases of the
nucleic acids).
- heteros (all
that is not amino-acid
or nucleic acid). A distinction is possible between solvent (water,
some
ions) and the other groups such as Hemes, lipids, etc.
- H bonds: a
facility to trigger the
display hydrogen bonds. You can specify to display hydrogen bonds
involving only 2 backbone or 2 side chain atoms, or hydrogen bonds
linking two different chains. Note:
There is no check of the consistency between the display of
the backbone or side-chains with the hydrogen bonds. Depending on the backbone and
side-chain display masks, hydrogen bonds may appear isolated from any
context.
- surface:
specify if you want or not a
representation of the molecular surface. The "interface" facility is
intended to
display the part of the surface located at the interface of two protein
chains.
Hence there is no interface for single chain entries. This "interface" feature is still experimental.
Also, nucleic acid chains are not considered currently. The
opacity of the surface can be adjusted to allow the remaining of
the structure to be seen by
transparency.
- More details
on the conventions used for the display and colouring patterns here
For
each of this
topics, you can choose a specific rendering mode, and
coloring pattern. I hope their names are rather self explicit.
Note: For each of the backbone,
side-chains, ... surface sections, you can choose None, in order to
adjust the representation using the supplementary
representations
Coloring
pattern notes:
- Charge: Presently, colouring is by residue type, no accurate
electrostatic potential calculatiuon is performed.
- Hydrophobicity: TRP,ILE,MET,VAL,TYR,LEU,PHE are considered as
hydrophobic. PRO and GLY are colored according to their amino-acid
colors. Other residues are coloured as hydrophylic.
- Class: Residue classes are: basic (ARG,LYS), acidic (ASP,GLU),
polar(SER,THR,TYR,HIS,CYS,ASN,GLN), aromatic (PHE,TYR,HIS,TRP),
aliphatic (ALA,GLY,ILE,LEU,MET,PRO,VAL).
- T factor: Colouring is a funciton of the values of the
temperature factors of the PDB file. Two zones are defined: low values
and large values. For each a color gradient is established. A
supplementary color gradient is established between the colors defining
respectively the end and the beginning ot the two zones. (See the advanced parameters section).
Drawing
modes:
- BallnStick: Atoms are displayed using small spheres, covalent
bonds are represented by cylinders joining the atoms
- Spheres: Atoms are displayed as spheres using as radius the Van
der Waals radii of the atomic types.
- Trace: Only the alpha-carbons are represented. Cylinders join the
consecutive alpha-carbons.
- Cartoon: A high level representation of the structure, involving
splines and information about the secondary structure. Beta strand
orientation is symbolized by arrows pointing towards the C-terminus.
- Lines: covalent bonds are represented by small cylinders joining
the atoms.
- Spline: The drawing followas a beta-spline passing through the
alpha-carbons.
- None: No display is performed.
ball and sticks
spheres
trace
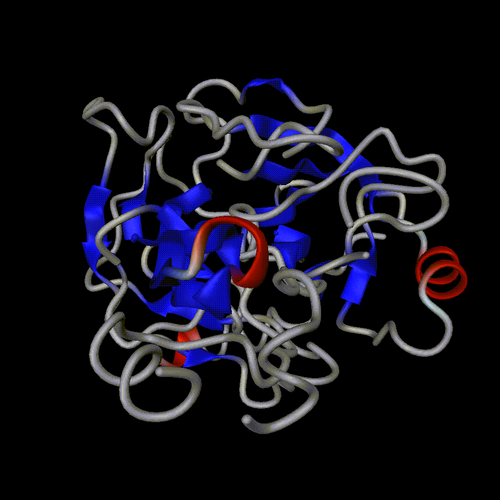
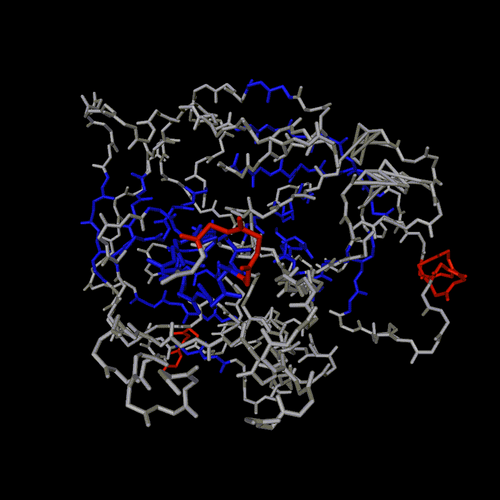
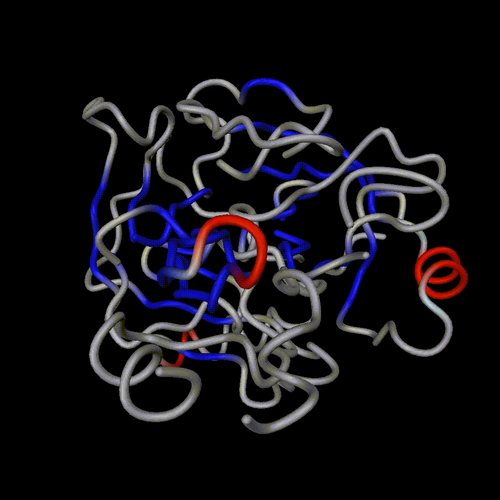
cartoon
lines
spline
Colouring patterns:
- Atom: Each atom is coloured depending on its type (carbon,
nitrogen, oxygen, sulfur, etc).
- Residue Type: The atoms of each residues are coloured depending
on the type of amino-acid (or base).
- Secondary structure: Parts of the structure that correspond to
alpha-helix, beta-strand or none of these are coloured
differently.
- T factor: The colours are assigned depending on the temperature
factor values specified in the PDB file. See the PDB
documentation for more explainations.
- Charge: Charges are assigned on the basis of negative charges for
ASP and GLU, positive charges for LYS and ARG. A more accurate
representation based on more realistic charges can be obtained using PCE-pot.
- Hydrophobicity: two classes of residues are coloured differently:
hydrophobic ("ALA,VAL,PHE,PRO,MET,ILE,LEU,TRP) and not hydrophobic
(remaining residues). In addition, PRO and GLY are coloured using the
colour of their residue types.
- Class: different colors are applied for the classes of residues:
basic (ARG,LYS), acidic (ASP,GLU), polar (SER,THR,TYR,HIS,CYS,ASN,GLN),
aromatic (PHE,TYR,HIS,TRP), aliphatic (ALA,GLY,ILE,LEU,MET,PRO,VAL).
- Chain: If the file contains several chains, each if coloured
differently.
- Named color: A unique colour is applied.
The colours associated with each of these patterns can be modified in
the advanced parameters section.
3.3 Scene parameters.
These parameters specify the parameters of the scene.
The default values will ensure that the protein is at the centre of the
image.
You can however specify precisely the orientation of the protein, by
filling the
fields "Centre on" and "Focus on". This will specify a line that goes
from the point specified as the centre to the eye of the user, passing
through the point defined as the focus. Additional rotations can be
applied (see the section Optional parameters).
- Centre on:
This specifies a coordinate that will be placed at the center of the
image. It can be on the form of the specification of a residue
name,
or atom name, or on the form of an explicit coordinate.
By default, the centre of mass is used. The specification of a residue
or atom must be on the form: C.RNI.A,
where C is a chain label
(1 letter), R, a residue
name (3letter code, 1 letter code possible for the 20 classical
amino-acids), N the PDB
file residue number (Important:
some PDB files do not start at residue 1. Here, we do you the numbers
of the PDB file. The sequence
visualization facility accessible from the form offers a mean to
check residue numbers), I
is the PDB insertion code (if any), A is an atom name (valid in the
residue specified). For example, A.ARG241B.CB denotes the carbon
beta of the arginine 241B (insertion code B) of the chain A. A.R241B.CB
would also be valid. Alternatively, you could edit the PDB file, pick
up the coordinates and specify for instance: 59.592
25.911 7.571 or 59.592 , 25.911 ,
7.571 (use a dot in numbers
and not commas, since commas will be removed).
- Focus on
:
specify a residue,
atom, or coordinate to place on a line that goes from the Centre
towards the eye of the user (Z axis). By default, the PDB file Z axis
is used.
- Advanced
parameters allow to specify rotations around X, Y and Z to adjust the
view.
- View
Angle
: specify the field of view angle of the camera for the picture
generation. As for a camera, the value should be positive. Reasonable
values are within the range 10..110. The "auto" value will trigger
calculation for a reasonable value.
- Stereo:
specify one mode to produce stereo images (using image
split). The default is no stereo, but you can choose one the straight
(right eye sees right image) or cross-eye (right eye sees left image)
scheme.
3.4 Adjusting structure orientation
This set of parameters allow to adjust the orientation of the
structure by small rotations (degrees).
The axes are defined as follows:
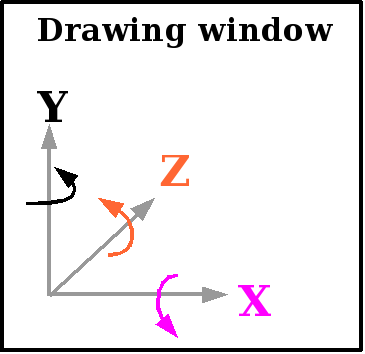
The X and Y axes are within the plane
of the drawing window, the Z axis is perpendicular to it. The arrows
indicate the positive rotations.
For a set of values, the rotations are applied to the structure
sequentially, following the order specified by the "order" option. Note
for a same set of values the resulting orientation will differ for
different rotation orders.
Since it can be difficult to assess the values to obtain some desired
orientation, a special facility designed for that purpose can be
accessed using the "preview orientation"
button. Once a satisfactory set of values has been identified, you can
simply report them from this help facility. Note that if you have
specified a "Focus" (see previous section), the rotations apply after
the focus transformation has been performed. If you modify the focus,
the rotation values will become meaningless.
3.4
Picture parameters.
- Background color,
Image size:
I
hope these are self explicit...
- Format: the
png and postscript
formats will produce a static image. The gif and mpeg formats will
result in animations. (see Animation).
- Animation:
4 types of animations
can be produced:
- Rock: the image will rock
around the Y axis.
- Xrot: the animation will
produce a full rotation around the X axis (horizontal)
- Yrot: the animation will
produce a full rotation around the Y axis (vertical)
- ZTtran: the animation will
produce a zoom into the molecule (eye-image center axis)
(Animation number of frames and step can be adjusted. See the advanced parameters sections).
- Produce:
The default (simple) is to generate a single image. Specifying
ortho2 or ortho3 will result in the production of 2 (resp.3) images
presenting different perpendicular views (Y and X
rotations).
4. Supplementary
representations:
This facility provides a mean of rendering some subparts of the
molecule using a representation different from the default
representation. Up to four independent selections
can be specified.
For each, you need to
specify the subset, its rendering mode, and its coloring pattern.
Note: For each selection, specifying
Display as: None will result in applying the colouring pattern on the
default representation.
Presently, although this might
evolve, selections correspond
to close to Dino valid selection expression, but are restricted, in
particular concerning keywords (see below).
Example to select the backbone of TYR and ARG within residues 25 to 58
and 70 to 92:
rname=TYR,ARG and rnum=25:58,70:92 and not aname=CA,N,C,O
Complex selections such as a selection of the form :
(rname=TYR,ARG and rnum=25:58,70:92) or (rnum=25:58,70:92 and
aname=CA,N,C,O)
i.e. a group of 2 elementary selections is also valid. Here, this
selects all TYR and ARG for residue numbers 25 to 58 and 70 to 92
and atoms CA,N,C,O (heavy atoms of the backbone) for residues 25 to 58 and 70 to 92.
Valid keywords:
protein, dna, rna
backbone, solvent,
hydrophobic,
basic, acidic, polar, aromatic, aliphatic
purin, pyrimidin, base
Example:
backbone and rnum=25:58 and aromatic
Note:
keywords are NOT used with "$".
Important Note:
to
avoid confusion, the "backbone" and "base" keywords correspond
respectively to the protein backbone atoms and the nucleic acids base
atoms. Hence, "protein and base" or "dna and backbone" lead to void
selections.
Use: "(protein and backbone) or (dna and not base)" for a selection of
the protein and nucleic acid backbone.
Use: "(protein and not
backbone) or (dna and base)" for a selection of protein side chains and
nucleic acid bases.
5.
Process:
This
will launch
the computation.
Depending on the complexity of the request and the server load,
it
might take from seconds
to several minutes.
The results will present the pictures (for
the formats supported by you browser), and a version of the script used
to render, but not preserving the file names, since these are
necessarily different on you computer and on the web server. No
acces to the MSMS files describing the molecular surface (if
necessary) is provided. You need to install
MSMS on your side to produce these.
6.
Advanced
parameters:
This section allows the customization of some of the
most important parameters. It is organized by groups of parameters.
- Title options: you can change the font, its size, the color of
the text, as well as the position of the first character of the text.
- Focus options: you can specify translation values
along the three axes. X is parallel to the top and bottom of the
screen, Y, to its edges, and Z points from the user's eye towards the
center of the picture. Translations are performed after teh
rotations, if specified.
- Animation options: These allow to specify the magnitude of the
animation. The move is rendered using a "number of Frames". Each of it
is the result of an elementary transformation depending on the nature
of the animation. The magnitude of this elementary transformation is
dependent on the "Step" for the Z translation and the Rock animation.
For the Z translation, the step corresponds to a displacement in
Angstroms along the Z axis. For the Rock animation, this corresponds to
a rotation in degrees around the Y axis.
- Color options: I hope these are self explicit. For the
temperature factors (TFac), the range of values is decomposed as three
zones: a lowvalue range (TFac1), an intermediate (not named) and a
large value range (TFac2). You can specify the values delimiting the
zones, as well as the colors at the boundaries. For the intermediate
zone, the colors at the boundaries correspond to TFac1ToColor and the
TFac2FromColor.
7.
Citations:
When publishing images
using surface rendering, please cite:
Sanner, M. F., Olson A. J. & Spehner, J.- C. (1996). "Reduced Surface: An Efficient Way to
Compute Molecular Surfaces." Biopolymers 38: 305- 320.
HBonds calculation:
I.K. McDonald and J.M. Thornton (1994), "Satisfying Hydrogen
Bonding Potential in
Proteins", JMB 238:777-793.
Secondary structure identification
using stride:
Frishman D, Argos P." Knowledge-based protein secondary
structure assignment." Proteins. 1995 Dec;23(4):566-79.
Dino:
DINO: Visualizing Structural Biology (2003) Ansgar Philippsen http://www.dino3d.org
8.
Feedback:
Please
send comments, suggestions, images to add to the gallery to: 
9. Gallery / examples:
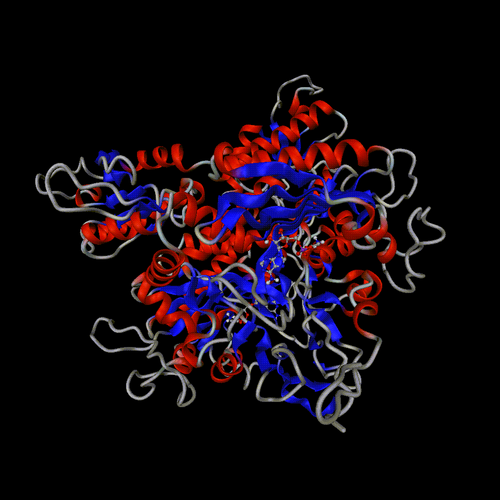
PDB entry 1ggm
default view
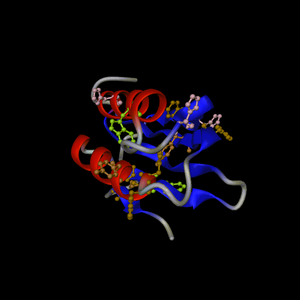
|
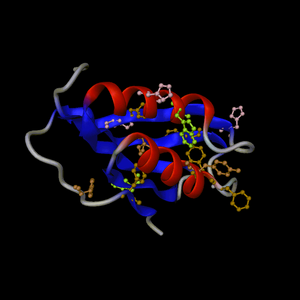
|
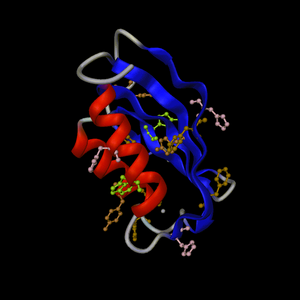
|
PDB entry
2acy: 3 orthogonal views.
Default representation supplemented by aromatic residues
coloured by residue type
|
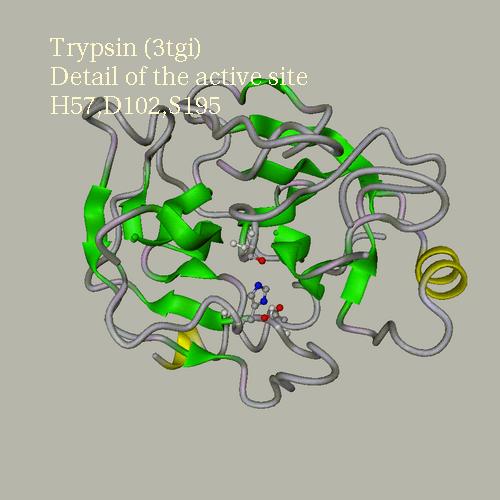
Example
of the trypsin active site. PDB entry is 3tgi.
Backbone default representation is set to "None". The focus is on
I.LYS15. The view angle is 30. An additional rotation of 30° on the
Y axis is performed. The title text is:
"Trypsin (3tgi)\nDetail of the active site\nH57,D102,S195"
Supplementary representations:
1: "chain=E" display as cartoon, color using secondary structure
2: "chain=E and rnum=57,102,195 and not backbone" color according
atomic types
3: "chain=E and rnum=57,102,195 and
backbone" color as "ivory"
The background is set to grey,
text color is lemonchiffon
Secondary structure colors are
green for strands, yellow for helices.
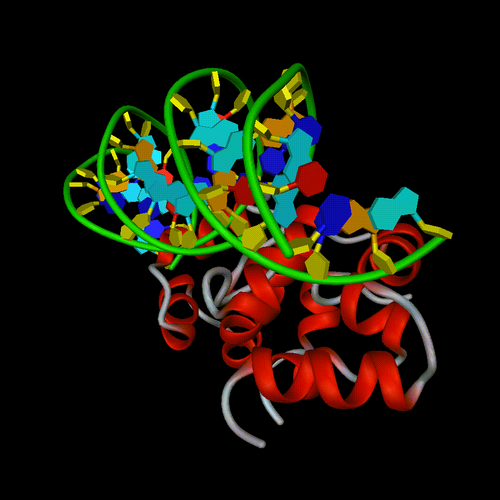
Example of
default representation of the 434 CRO
PROTEIN COMPLEX WITH 20 BASE PAIR PIECE OF DNA CONTAINING OPERATOR OR1
(PDB entry 3cro).
Default parameters. View angle = 30
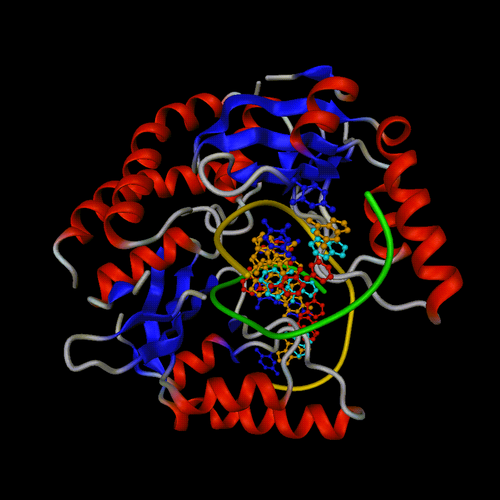
Example of combining only supplementary representations on the 1eyu PDB
entry.
All default representations set to None. Rotation on X by -30 degrees.
Supplementary representation 1 set to: protein, displayed as cartoon,
coloured by secondary structure.
Supplementary
representation 2 set to: dna and not base, displayed as spline,
couloured by chain (chain D - 4th chain - using gold).
Supplementary
representation 3 set to: dna and base, displayed as balls
and sticks, couloured by residue type.
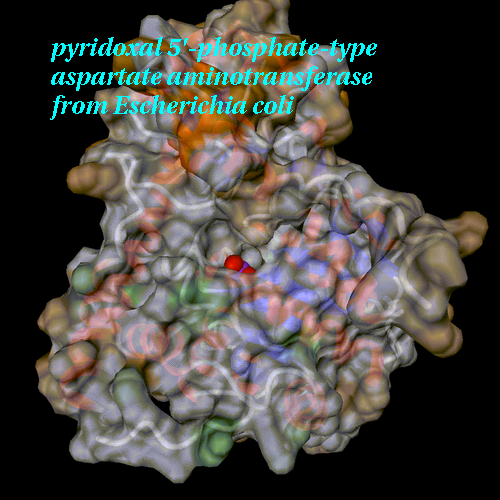
PDB entry 1art,
surface with opacity 0.75 coloured according to temperature
factors
The default backbone representation is preserved.
Heteros groups are displayed as spheres, coloured by atomic types.
View angle set to 30, additional rotation around X by 30° .
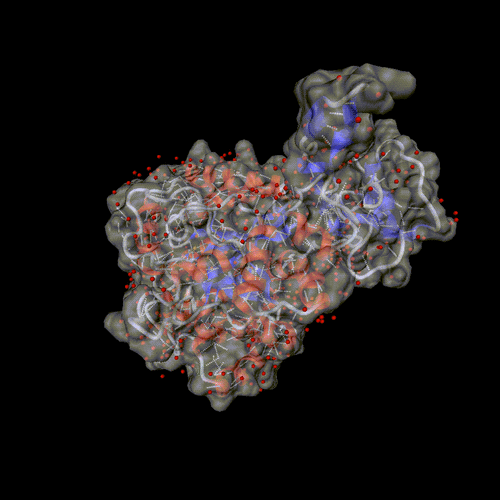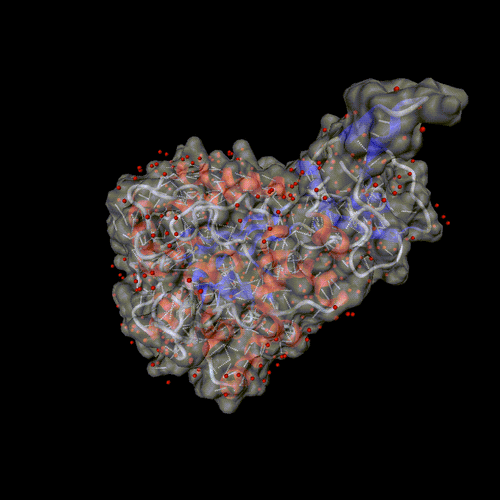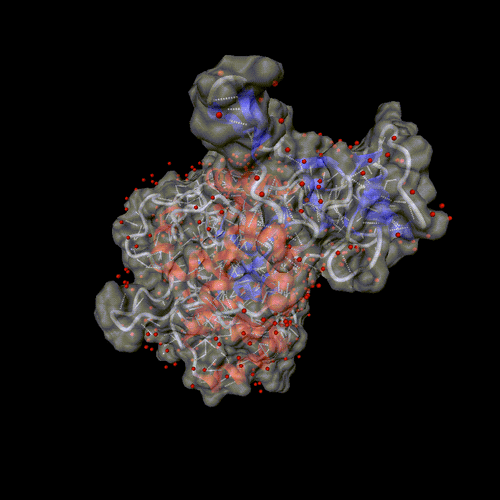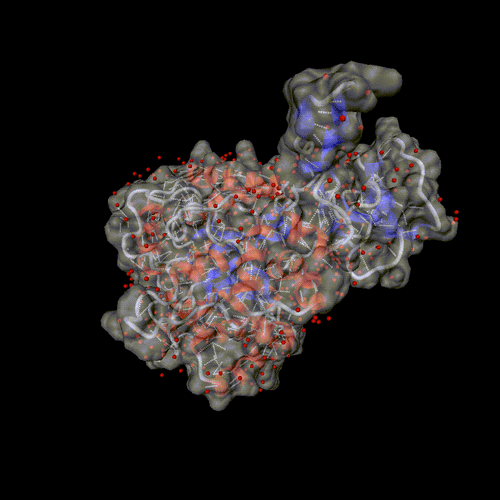
Cytosine deaminase.
PDB entry 1rak.
The solvent is displayed around the molecular surface, transparent to
show the backbone and the hydrogen bonds.
Optional parameters: Surface, and solvent set to all.
Image format: gif, animation: rock
All other parameters to their default values.

Mechano sensitive channel.
The view is on the axis of the channel.
Centre is set to: 27.498 128.966 7.763e-06 (centre of mass)
Focus is on: 25.966 127.680 8.459e-06 (displacement along the principal
inertia axis of the structure)
The surface is coloured according to Temperature factors
Gif format. The animation is a translation along Z
Thanks to C. Etchebest and F. Guyon.

Comparing two sets
of aromatic side chains conformations.
No defaul display. The input file was generated by concatenating two
PDB files, assigning each a chain label (A and B).
selection1 set to "chain = A and not backbone and (aromatic)", coloured
by residue type
selection2 set to "chain = B and not backbone and (aromatic) ",
coloured as grey