Overview
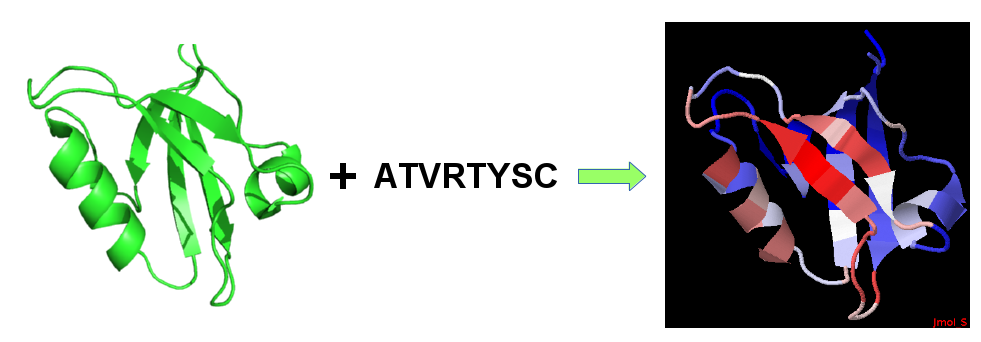
Protein structure and peptide sequence to identify candidate sites of interactions on protein surface.
Why PEP-SiteFinder ?
The recent years have seen increased interest for peptides to probe molecular interactions, or for the early design of candidate therapeutics, which makes higly desirable the detailled caracterization of the interaction between the peptides of interest and their target proteins. In silico docking approaches have shown efficient to generate conformations of the complexes but remain however strongly dependent on the preliminary knowledge of the region of the protein involved in the interaction and, to some extent, on that of the peptide conformation. The blind indentification of candidate regions of the protein surface likely to be involved in the interaction with a peptide is thus often required to assist the design of in vitro or in silico experiments. PEP-SiteFinder is a service designed to assist such caracterization.
Run PEP-SiteFinder
The
PEP-SiteFinder service is integrated in the
RPBS Mobyle Portal.
When using this service, please cite the following reference:
Saladin A, Rey J, Thevenet P, Zacharias M, Moroy G, Tufféry P.
PEP-SiteFinder: a tool for the blind indentification of peptide binding sites on protein surfaces.
Nucleic Acids Res. 2014 Jul;42(Web Server issue):W221-6.
Latest news
- March, 11th, 2014: Updated benchmark results.
- February, 5th, 2014: Implemented a filter for the early detection of PDB input containing nucleic acids, that are not presently supported.
- January, 29th, 2014: PEP-Site at CAPRI round 29, target 66: PEP-SiteFinder targeted the right patch !
- January, 21th, 2014: Added a pymol script for local visual inspection of the results.
- January 20th, 2014: Slight speed up of the processing (~20%). Average processing time on the order of 25 mn, depending on complex size, and server load. Very large systems can however require over 2 hours.
- January 17th, 2014: Added a plot illustrating the relationship between the propensities and the probability that the site is at the protein-peptide interface.
- January 16th, 2014: Added a note about the benchmark dataset.
- January 15th, 2014: Better management of multiple chain proteins.
- January 14th, 2014: Simpler interface for the interactive visualization of the results.
- November 22, 2013 : Benchmark against the peptidb dataset.
- October 10, 2013 : Service opens.
PEP-SiteFinder' design
PEP-SiteFinder identifies candidate regions for the protein-peptide interactions by performing series of blind docking experiments in which the protein and the peptide conformations are rigid, which makes the search rather fast (few tens of minutes).
PEP-SiteFinder relies on two main components that are:
- PEP-FOLD, an approach to the fast generation of peptide conformation. It is a de novo approach aimed at predicting
peptide structures from amino acid sequences. This method,
based on the concept of structural alphabet (SA) that is a kind of generalized secondary structure, where the SA "letters" describe the
conformations of four consecutive residues (see [1] ). PEP-FOLD predicts for each fragment of four reisudes in a query sequence the most probable local conformations - SA letters -. It then performs the 3D assembly of the complete structure using a greedy algorithm and the sOPEP
coarse-grained force field (see [2] ). PEP-FOLD latest evolution improves performance for linear peptides up to 36 amino acids - best model with an averaged RMSd of 2.1 A from NMR structure ( [3] ).
- PTools , a tool for the generation of protein protein complexes, for which specific parameters for protein-peptide interactions have been learnt. PTools performs a blind rigid body search: the conformation of the partners is kept rigid during the conformational search, which is achieved by the systematic scan of the protein surface to generate poses that are refined using a simple minimization approach. The minimization process and the scoring of the poses relies on the coarse grained force field ATTRACT. (see [4] , [5] ))
Given a peptide sequence, PEP-SiteFinder first runs PEP-FOLD to generate collections of sub optimal conformations which are clusterized. A maximum of 20 representative conformations is selected. Some variations in the conformations selected can occur from a run to another. Each conformation is then used in turn to generate protein-peptide complexes using PTools. The best poses are scored using ATTRACT2, and we have found that such procedure results in identifying complexes in which few patches of the protein surface are involved, most often encompassing the experimental region of interaction.
What is PEP-SiteFinder not ?
PEP-Site finder is a service to identify candidate patches on a protein surface with which a peptide of specified sequence is likely to interact. It does not provide reliable information about the energies of interaction. It presupposes that the peptide binds to the protein.
PEP-SiteFinder relies on a fast approach to generate peptide conformations in solution . The conformation of the peptide in the complex might be different from it. However, we have observed that sub optimal conformations generated for the peptide alone are concistent with the identification of the correct protein patches involved in the interaction with the peptide, using a rigid docking approach.
Note that the complexes returned are usually a rough approximation of the actual complex structure. It has for instance been observed that the patches identified can be correct despite a flip of the peptide, or despite the peptide conformation does not correspond to the experimental conformation in the complex. For users interested in a detailled caracterization of the protein peptide interaction, PEP-SiteFinder complexes should only be considered as a starting point for more focused docking.
Features
-
Candidate patch identification:
Starting from a single amino acid sequence, PEP-FOLD improved version runs up to 200
simulations and identifies a maximum of 20 candidate conformations, the centroids of the 20 best clusters according to sOPEP.
For each peptide conformation, a blind search is performed and the 10 best poses are selected. A maximum of 200 poses are thus selected, and sorted according to their ATTRACT2 sccores.
-
Residue interaction propensities:
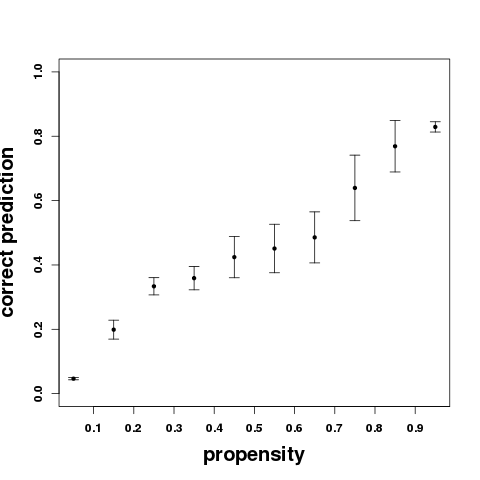
An interaction propensity index - percent times the residue was contacted in the 50 best poses - is calculated for each residue of the protein, from the selected poses. This index is correlated with the probability that the residue is at protein-peptide interface. Residues with propensity values more than 0.7 have a probabiliy over 0.8 to be at the interface. (see benchmark)
-
Graphical exploration of the complexes:
The poses can be explored thanks to the Jmol applet ( [6] ). Note this requires a Java plugins installed in your browser.
-
Coordinates of the complexes are available for further off-web exploration:
Two files in the PDB format are available. The first corresponds to the protein, in which the Temperature factors fields
are replaced by the interaction propensity values. The second one corresponds to the peptide, and the multiple poses are returned
using the MODEL facility of the PDB format. The models are sorted according to their ATTRACT2 scores, MODEL1 being the model with the best score, etc.
Limitations
Usage
The input consists in two fields to specify the protein structure and the peptide sequence.
-
Protein structure: It corresponds to the structure of the protein in interaction with the peptide. It must be in the PDB format. Note that PEP-SiteFinder does not presently accept input other than proteins. The presence of nucleic acid chains for instance, will not be accepted. Also note that HETATM lines are discarded during the docking process.
-
Peptide sequence: The amino acid sequence of the peptide interacting with the protein. It must be specified in the
FASTA format. The
query peptide sequence must contain a string of only the 20 standard
amino acids in uppercase, using the 1 letter code (see the pre-configured test example).
The size of the input sequence can be as long as 30 amino acids, and longer than 5 amino acids which is a lower limit for the generation of 3D conformation using PEP-FOLD.
-
Pre-configured test: By setting this option to "Yes", PEP-SiteFinder will run a test case for a GRIP1 PDZ domain in complex with liprin C-terminal peptide in interaction with the 8 mer peptide "ATVRTYSC", i.e. the peptide sequences corresponding to the chain D of 1N7F. The protein coordinates are that of the APO structure of the protein (PDB code: 1N7E chainA, residues 668-753) corresponding to the residues of complexed structure (PDB code: 1N7F). The results can be compared with the coordinates of the complex (PDB code: 1N7F).
By switching the demonstration mode, other input data specified in the other fields will be ignored.
Results
-
Progress report

This section will incrementally provide information about job progression and errors if any.
A typical run should produce a report similar to that. Errors related to the input data specified are now also reported in this field.
-
Note typical PEP-SiteFinder runs are on the order of 30 minutes , depending on the size of the protein, and the number of peptide conformations generated using PEP-Fold.
-
An interactive page allowing to browse the best complexes generated and explore the residues having large propensities of interaction, varying the representations of the protein and peptide, and displaying the protein residue propensities to be at the interface.
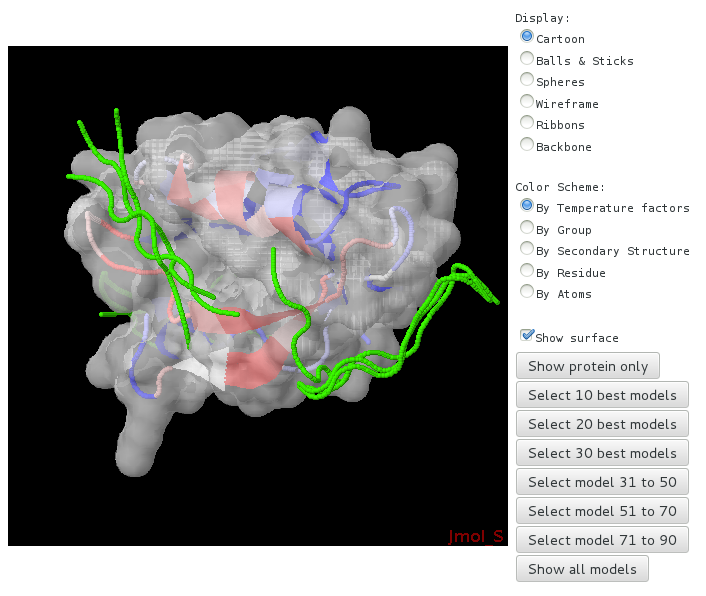
- PDB with the interaction propensitites set in the temperature factor field.
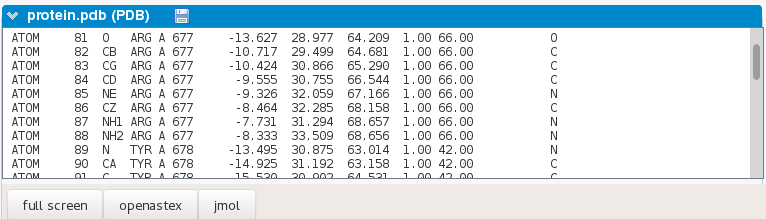
-
PDB of the poses, organized as a multiple MODEL PDB, where models are sorted according to their ATTRACT2 score.
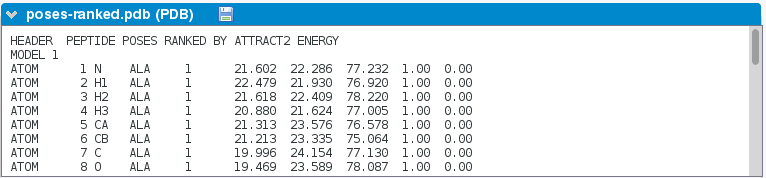
-
Per residue propensities list.

-
Page of downloadable files.
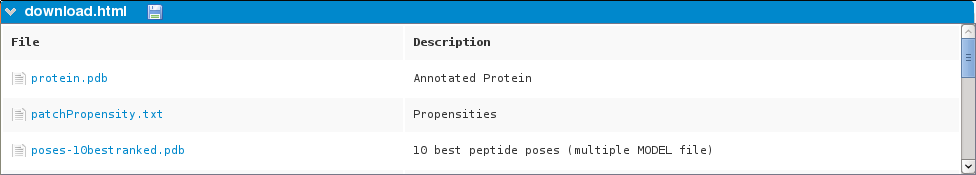
The following files are available:
- Protein PDB file
- Propensities
- PDB (multiple model) file with the 10 best poses ranked by energy
- Pymol script to load the 10 best poses:
Pre-selections include different items such as protein (with surface on), peptide poses loaded as different "states" inside an unique object ("ranked"), and the different peptide poses ranked by energy (ranked_0001, ranked_0002, etc...).
- PDB (multiple model) file with the 50 best poses ranked by energy
- Pymol script to load the 50 best poses
- PDB (multiple model) file with all the poses ranked by energy
- Pymol script to load all poses
- Archive (tgz) containing PDB files of all the poses sorted by conformation
- Pymol script to load all poses by peptide conformation (archive poses.tgz must be extracted):
Pre-selections include items such as protein (with surface on) and the different peptide conformation (cnf1, cnf2, etc...) with each pose loaded as different "states".
Examples
HDM2 alpha-helical transactivation domain of P53 (PDB code: 1N7F):
|
|
|
|
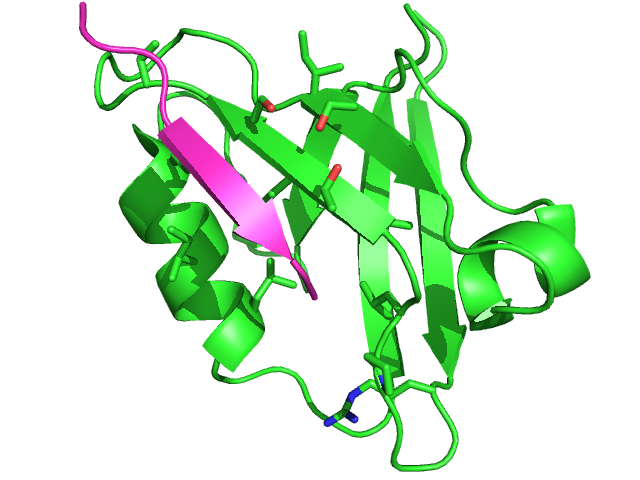
Experimental complex structure (1N7F)
green: protein; cyan: peptide
|
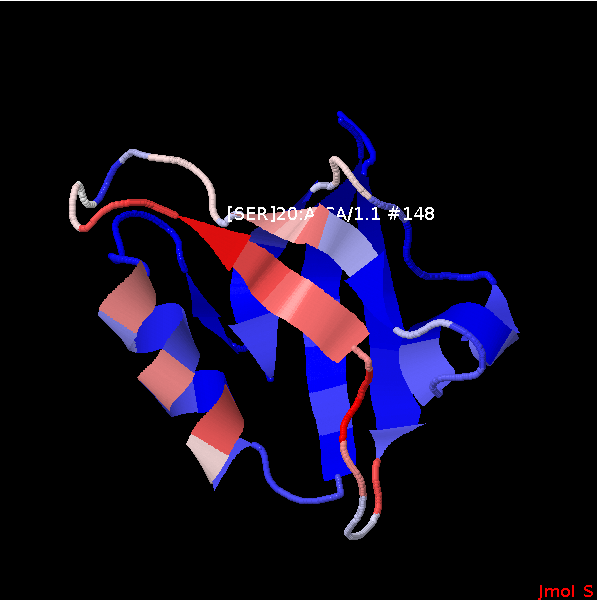
The APO structure (1N7E) coloured according to residue propensities returned by PEP-SiteFinder.
(red, large values; blue, smallvalues).
|
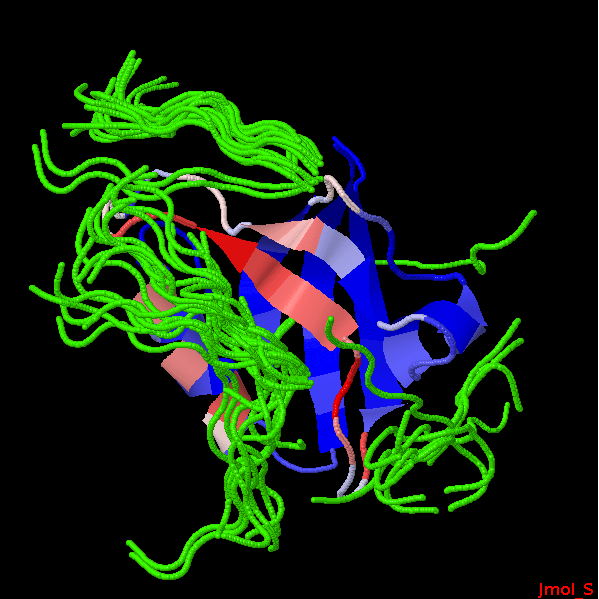
The 50 best peptide poses generated by PEP-SiteFinder to derive the propensities.
|
The hotest sites corresponds to PRO14, LEU15, and SER20, residues close to or in contact with the ligand in the experimental complex. The strand including SER20 and the helix including LEU65, ILE69, LEU72 define a region with large propensities that corresponds to the region of the actual experimental interaction. The side chains identified by PEP-SiteFinder with propensities more than 40% are detailled in the experimental structure of the complex (left).
The interactive page generated by PEP-SiteFinder is accessible here .
This page requires that the Java plugins is active in your browser. You can use the buttons to change representation, and to select the poses, ranked according to their ATTRACT2 scores. The propensities are available here .
Human MDM2 in complex with an optimized p53 peptide (PDB code: 1T4F):
|
|
|
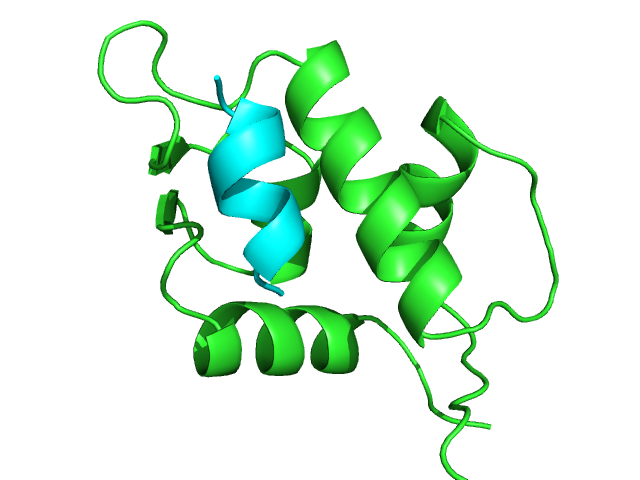
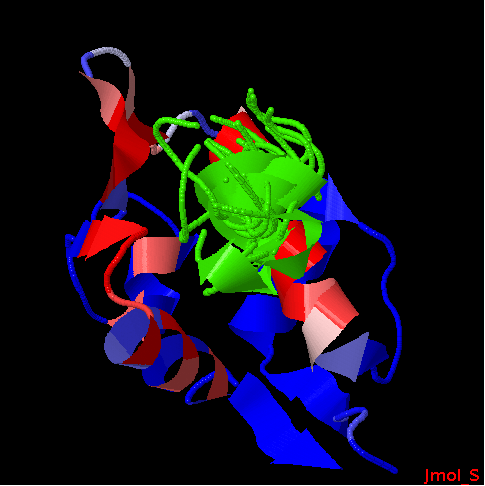
Experimental complex structure (1T4F)
green: protein; cyan: peptide
|
10 best peptide poses generated by PEP-SiteFinder.
|
The interactive page generated by PEP-SiteFinder is accessible here .
This page requires that the Java plugins is active in your browser. You can use the buttons to change representation, and to select the poses, ranked according to their ATTRACT2 scores. The propensities are available here .
CAPRI round 29, target 66: PriA Helicase Bound to SSB C-terminal Tail Peptide (PDB code: 4NL8):
|
|
|
|
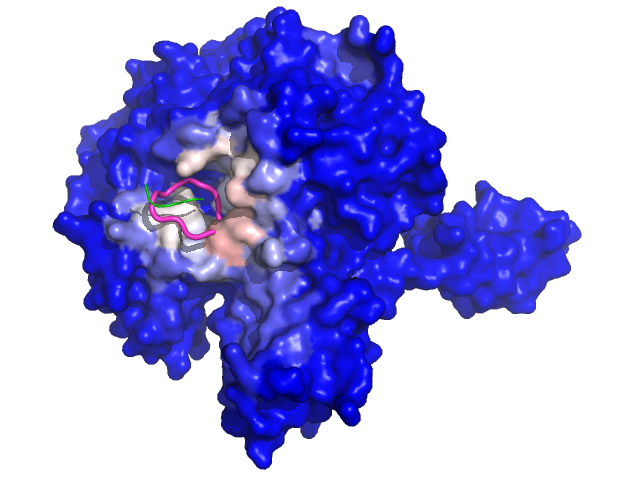
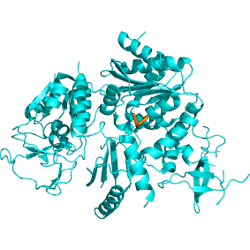
Experimental structure |
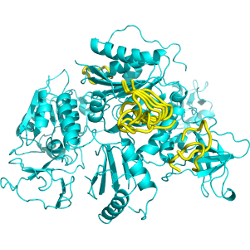
10 best peptide poses generated by PEP-SiteFinder |
CAPRI 29 complex structure is out ! The best spot returned by PEP-SiteFinder corresponds to the actual interaction site. The experimental structure, shown in cyan, only details the five last residues of the peptide, shown in orange (middle). The 10 best poses of the complete peptide (10 amino acids) generated by PEP-SiteFinder is depicted (right).
Benchmark
Dataset: The performance has been assessed on 41 high-resolution peptide-protein complexe of the PeptiDB dataset (
[7] ) for which the structure of the free protein (uncomplexed) is available. These complexes have been selected, starting from the complete PeptiDB collection of 103 complexes on the basis of the peptides size - 5 to 15 residues long, of the resolution of crystal structure - resolution better than 2 Å, and on the absence of any heteroatoms at the interface. To prevent redundancy, the sequence identity, below to 70%, and the difference of the protein foldings have also been taken into account.
Note: the small size of the collection of protein-protein complexes used for benchmark is very similar to the size of the subset that would be extracted from the PepSite ( [8] )collection of over 400 complexes, selecting the complexes resolved at neutral pH, at a sufficient resolution (2.5 A or better) and R value (0.3 or lower), and considering peptides with only standard amino acids, which results in only 48 complexes, not considering protein redundancy.
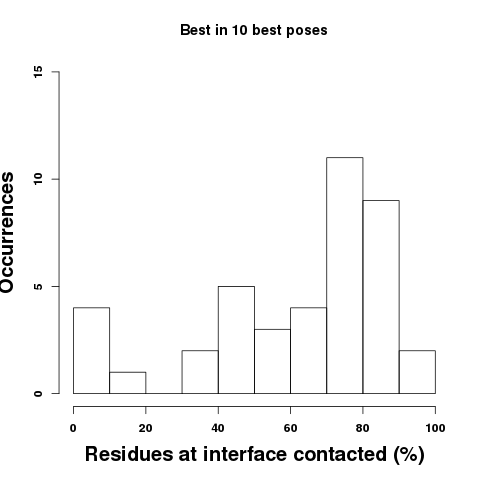
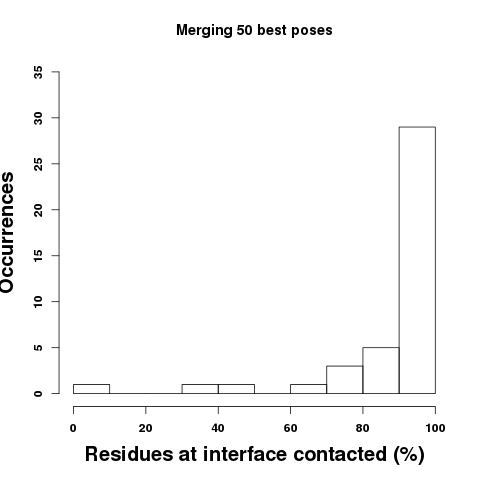
Results: Top: Considering the 10 best poses, we find that the best pose is able to overlap to some extent residues of the binding site in all cases but 4 (10%). On average, PEP-SiteFinder best pose among 10 is able to identify more than 50% of the protein residues contacted by the peptide for more than 70% cases. The residues contacted are identified considering heavy atoms only, using a cutoff of 5 angstroms to identify atoms in contact.
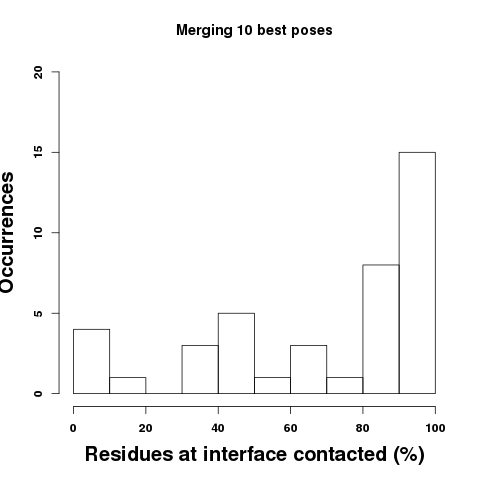
Bottom: Considering the information that can be derived combining the 10 or 50 best poses, one sees a shift of the amount of residues at the interface that are contacted. Considering the 50 best poses, the fraction of residues of the binding site contacted by at least one pose is on average of 90%. Following, we find that the probability that a residue is effectively at peptide interface (correct prediction) increases when the propensity values increase, as illustrated by the bottom right plot. The propensities thus provide information about the confidence of the prediction. Note the results can vary a bit depending on the 3D conformations of the peptide generated by PEP-FOLD. The error bars correspond to the standard deviation observed over 5 independant runs.
References
[1] Camproux AC, Gautier R, Tuffery P.
A hidden markov model derived structural alphabet for proteins.
J Mol Biol. 2004 Jun 4;339(3):591-605.
[2] Maupetit J, Derreumaux P, Tuffery P.
A fast and accurate method for large-scale de novo peptide structure prediction.
J Comput Chem. 2010 Mar;31(4):726-38.
[3] Thevenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tuffery P.
PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides.
Nucleic Acids Res. 2012. 40, W288-293.
[4] Saladin A, Fiorucci S, Poulain P, Prevost C, Zacharias M.
PTools: an opensource molecular docking library.
BMC Struct Biol. 2009 May 1;9:27.
[5] Schneider S, Saladin A, Fiorucci S, Prevost C, Zacharias M.
ATTRACT and PTools: open source programs for protein-protein docking.
Methods Mol Biol. 2012;819:221-32
[6] Hanson R.M.
Jmol - a paradigm shift in crystallographic visualization.
Journal of Applied Crystallography, 2010, 43(5): 1250-1260
[7] London N., Movchovitz-Attias D. and Shueler-Furman O.
The structural basis of peptide-protein binding strategies.
Structure, 2010, 18(2): 188-199
[8] Petsalaki E, Stark A, Garcia-Urdiales E, and Russell RB.
Accurate prediction of peptide binding sites on protein surfaces.
PLoS Comput Biol. 2009, 5(3):e1000335.