Welcome to the PEP-FOLD 2011 improved service!
PEP-FOLD is a de novo approach aimed at predicting peptide structures from amino acid sequences. This method, based on structural alphabet SA letters to describe the conformations of four consecutive residues, couples the predicted series of SA letters to a greedy algorithm and a coarse-grained force field.
What's new:
Jan 2016: PEP-FOLD3 is on-line. Faster, open for linear peptides in solution from 5 up to 50 amino acids, allows preliminary peptide protein interaction studies. Not optimized yet for disulfide bonds or user specified constraints. Use this service for such constrained peptides.
PEP-FOLD latest evolution improves performance for linear peptides up to 36 amino acids - best model with an averaged RMSd of 2.1 A from NMR structure, also allows user specified constraints such as disulfide bonds and inter-residue proximities.
 |
 |
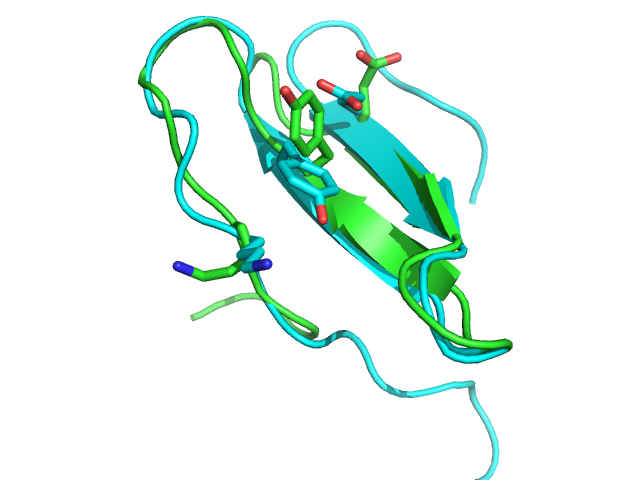 |
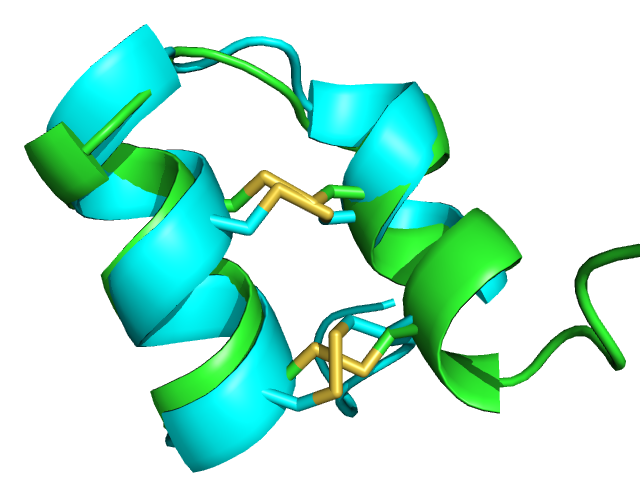
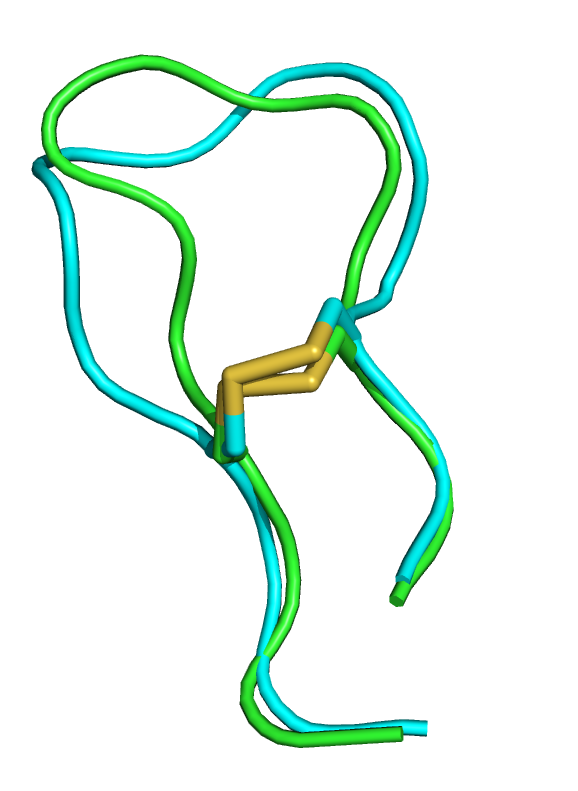
| 1wqc PEP-FOLD best model (cyan) and experimental (green) conformations. 1.4 A RMSd. | 1jbl PEP-FOLD best model (cyan) and native (green) conformations. 1.8 A RMSd. | 1e0n PEP-FOLD best model (cyan) and experimental (green) conformations. Some side chains illustrate the beta sheet superimposition. |
Access the
PEP-FOLD server @ the
RPBS Mobyle Portal.
When using this service, please cite the following references:
Improved PEP-FOLD approach for peptide and miniprotein structure prediction
J. Chem. Theor. Comput. 2014; 10:4745-4758
PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides.
Nucleic Acids Res. 2012. 40, W288-293.
Older references:
PEP-FOLD: an online resource for de novo peptide structure prediction.
Nucleic Acids Res. 2009. 37(Web Server issue):W498-503. doi:10.1093/nar/gkp323
A fast and accurate method for large-scale de novo peptide structure prediction.
J Comput Chem. 2010. 31-726-38.
[ Presently restricted to peptide sizes from 9 to
36 residues ]
When using this service, please cite the following references:
Improved PEP-FOLD approach for peptide and miniprotein structure prediction
J. Chem. Theor. Comput. 2014; 10:4745-4758
PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides.
Nucleic Acids Res. 2012. 40, W288-293.
Older references:
PEP-FOLD: an online resource for de novo peptide structure prediction.
Nucleic Acids Res. 2009. 37(Web Server issue):W498-503. doi:10.1093/nar/gkp323
A fast and accurate method for large-scale de novo peptide structure prediction.
J Comput Chem. 2010. 31-726-38.
New Features
- Peptide structure prediction: Starting from a single amino acid sequence, PEP-FOLD improved version runs up to 200 simulations rather than 40 before. By default, 100 simulations are performed. It returns an archive of all the models generated, the detail of the clusters and the best conformation of the 5 best clusters.
-
Prediction constraints: The
PEP-FOLD accepts user specified constraints such as disulfide bonds or residue proximities.
Note that constraint satisfaction is not systematic in all the models returned since (i) constraints - particularly if erroneous - can, in some cases, conflict with the local structure prediction. (ii) PEP-FOLD is a coarse grained approach and the all atom generation from coarse grained representation can result in unclosed disulfide bonds in the all atom representation, even if the cysteines are close.
Important: PEP-FOLD does not perform disulfide bond prediction. You can guess disulfide bond connectivity from services such as: EDBCP
Other Features
- Reference structure: PEP-FOLD server allows you to upload a reference structure in order to compare PEP-FOLD models with it (see usage).
-
Fast folding: Execution time on the server usually vary from few minutes to less than one hour, once your job is
running, depending on server load. Usually, PEP-FOLD
prediction takes about 40 minutes for a 36-residue
peptide.
Note that PEP-FOLD prediction and folding is presently assumed for neutral pH.
Limitations
- Amino acid sequence size: For now, PEP-FOLD prediction is limited to amino acid sequence between 9 and 36 residues, although PEP-FOLD has been tested off-line for sizes up to 50 amino acids. For sizes more than 30 amino acids, users can contact the authors. We consider to open up to 50 amino acids depending on server load.
- Peptide properties: Presently, PEP-FOLD is not able to process peptides with D-amino-acids, non standard amino acids, nor circular peptides involving other than disulfide bonds.
Usage
Input
-
Pre-configured test: By setting this option to "Yes", PEP-FOLD will be run using the amino acid sequence of 1jbl:
>1jbl
GRCTKSIPPICFPD
specifying a disulfide bond between cysteine 3 and 11. All input data specified in the other input fields will be ignored. - Input sequence: Input sequence file must be in FASTA format. The query peptide sequence must contain a string of only the 20 standard amino acids in uppercase, using the 1 letter code (see the pre-configured test example). The size of the input sequence can be as long as 50 amino acids, but the simulation must be restricted to 36 amino acids using the "3D reconstruction range" option (see below).
Input options
- Run label: A title for your run. It MUST be a single word (no spaces, no special characters). It will be used to generate the name of the models.
- Type of simulation: The default short simulations correspond to 100 model generation runs. In most cases, our results show it is sufficient to identify the correct fold. Long simulations will launch the generation of 200 models.
- Sort models by: Once generated, models are clustered using Apollo [6] to identify groups of similar models. The clusters are then sorted using either the sOPEP energy value, or Apollo predicted TMscore (tm). For peptides up to 36 residues, using sOPEP as a key to to sort the clusters will often result in proposing native or near native conformations in the top 5 ranks.
- 3D reconstruction range: PEP-FOLD accepts sequence input up to 50 amino acids, but specifying a maximal 36-residue region subjected to 3D modelling. This facility is useful for peptides where the N- and C- terminal regions are known to be disordered and the user wishes to focus on the modelling of the structured core. To this end, our tests show that the prediction of the SA profile is best using the complete sequence rather than a truncated sequence. The region that will undergo the 3D modelling must be specified on the form: x-y (e.g. 15-50), indicating that the 3D models will encompass the region between residues 15 and 50 (included), where 1 is the first residue of the input sequence.
-
Disulfide bonds: You can specify the pairings of the cysteines using the format: x-y where x, y are the numbers of the cysteines, 1 being the number of the first residue in the sequence.
Multiple disufide bonds can be specified by separating pairings using a ':'. For instance 1-30:5-10 stands for 2 disulfide bonds. The first is between cysteines 1 and 30, the second between cysteines 5 and 10. - Residue proximities: You can specify one or several proximities between two residues using the same format as for disulfide bonds.
- Reference structure: Input reference structure file must be in PDB format. This file must only contains a single model, with no hetero-residues and have the exact same length (in amino acids) as the query sequence. If a 3D reconstruction range is specified, the reference structure must match exactly and *ONLY* the sequence for which 3D models will be generated. The residue number must match those of the models, i.e. start at residue 1 if no 3D reconstruction range is specified or at residue x if a range x-y is specified.
Results
- PEP-FOLD produces informative report to guide the user through the best model selection.
-
Progress report
 This section will incrementally provide information about job progression and errors if any.
A typical run should produce a report similar to that. Errors related to the input data specified are now also reported in this section.
This section will incrementally provide information about job progression and errors if any.
A typical run should produce a report similar to that. Errors related to the input data specified are now also reported in this section.
-
Clustering report
 This report is primarily a table file that describes the clusters. For each model generated, up to 10 numbers are reported:
avg, gdt, max, q, tm are the scores predicted by Apollo
[6], where gdt is the predicted GDT_TS,
q is the predicted Qmean score and tm is the predicted TMscore.
sOPEP is the coarse grained energy of PEP-FOLD. If a reference file has been specified in input,
additional columns correspond to the TMscore and the cRMSd obtained by aligning respectively the model onto the reference using TMalign or a
rigid fit procedure minimizing the alpha carbon RMS deviation. The CGCNS and nSS are related to user constraints. CGCNS stands for a Coarse Grained CoNstraint Satisfaction, between 0 and 100, where 100 means all the constraints satisfied. nSS stands for the number of disulfide bond obtained at the all atom representation level (N/A for not appliable).
This report is primarily a table file that describes the clusters. For each model generated, up to 10 numbers are reported:
avg, gdt, max, q, tm are the scores predicted by Apollo
[6], where gdt is the predicted GDT_TS,
q is the predicted Qmean score and tm is the predicted TMscore.
sOPEP is the coarse grained energy of PEP-FOLD. If a reference file has been specified in input,
additional columns correspond to the TMscore and the cRMSd obtained by aligning respectively the model onto the reference using TMalign or a
rigid fit procedure minimizing the alpha carbon RMS deviation. The CGCNS and nSS are related to user constraints. CGCNS stands for a Coarse Grained CoNstraint Satisfaction, between 0 and 100, where 100 means all the constraints satisfied. nSS stands for the number of disulfide bond obtained at the all atom representation level (N/A for not appliable).
Additionally, this table gives access to model download at different levels: The archive of all the models (top of table), archives of all models in a cluster - if the cluster contains several models, and each individual models.
The cluster ranks are defined according to their scores (sOPEP or tm) - see the input option "Sort models by". The cluster representatives correspond to the models of the clusters having the best scores, i.e. with the lowest sOPEP energy (resp. highest tm value). They are denoted as "modelx", where x is the rank of the cluster according to the sort key . When a cluster has several models, it is in turn sorted according to the sort key. The first model in the table is the representative, denoted on the form "modelx" and the following models are numbered using the "modelx.y" convention where x is the rank of the cluster and y the rank of the model in the cluster.cRMSd* (alpha carbons Root Mean Square deviation), GDT_TS* (Global Distance Test Total Score, for more information see: predictioncenter.org), sOPEP energy (see concepts section), TM score* (for more information see Zhang server) and a single character indicating if it is the cluster centroid (*) or not (-).
* compared to the reference structure if provided. -
Model 1 to 5Representatives o the 5 best clusters (according to tm - see CLustering report) predicted structure are provided in PDB format.
 You can either save the file onto your computer, or view it using JMol.
In the Mobyle environment, PDB files can also be piped to other analyses such as the identification of seondary structures using stride or p-sea.
For this, select the approriate method beside the "further analysis" button, then auch it by clicking on "further analysis".
You can either save the file onto your computer, or view it using JMol.
In the Mobyle environment, PDB files can also be piped to other analyses such as the identification of seondary structures using stride or p-sea.
For this, select the approriate method beside the "further analysis" button, then auch it by clicking on "further analysis".
-
Local Structure prediction profile
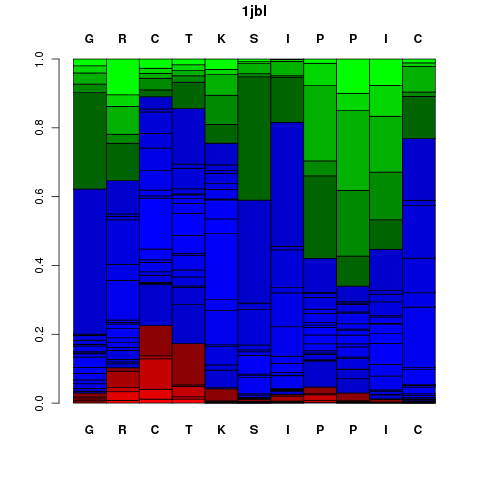 It corresponds to a graphical representation of the probabilities of each Structural Alphabet (SA) - see the
Concepts-
letter (vertical axis) at each position of the sequence (horizontal axis). Note that SA letters correspond to fragments of 4 residue length.
The profile is presented using the follwing color code: red: helical, green: extended, blue: coil.
It corresponds to a graphical representation of the probabilities of each Structural Alphabet (SA) - see the
Concepts-
letter (vertical axis) at each position of the sequence (horizontal axis). Note that SA letters correspond to fragments of 4 residue length.
The profile is presented using the follwing color code: red: helical, green: extended, blue: coil.
-
Models archive
 This archive contains all the models generated. It is in the unix tar format compressed using gzip.
This archive contains all the models generated. It is in the unix tar format compressed using gzip.
Once saved on your computer, enter for instance (unix) tar xzf AllModels.tgz to inflate the archive.
Examples, sample tests
-

A Mobyle video tutorial is available [WMV format (14 Mb)]. - As a simple test, you can either choose to:
-
Copy, paste the following
sequence to the "Peptide amino acid sequence" field:
>1egs_A mol:protein length:9 GROES
TKSAGGIVL - or use Mobyle facilities :
-
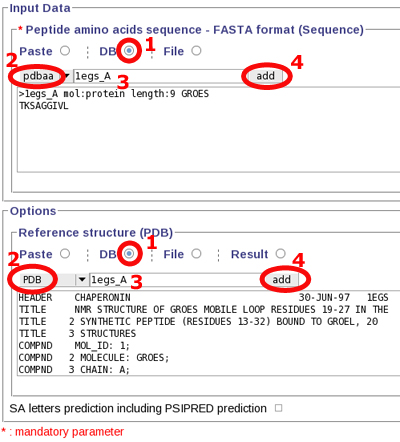 Fill the input data 1. click
"DB" radio button 2. select pdbaa database 3. write your
sequence identifier (here: 1egs_A) 4. add this sequence
to the form field,
Fill the input data 1. click
"DB" radio button 2. select pdbaa database 3. write your
sequence identifier (here: 1egs_A) 4. add this sequence
to the form field,
- Fill the options 1. click "DB" radio button 2. select PDB database 3. write your sequence identifier (here: 1egs_A) 4. add this PDB file to the form field.
- In both cases:
-
Run Launch PEP-FOLD
prediction, by clicking "Run" at the top of the page.
nota bene: For non-registred users, a captcha will ask you to type the text from the image before submitting your job. Once your job has been submitted, you can check your results availability by clicking the "update job status" button.
Concepts
- Structural alphabet PEP-FOLD is based on the concept of structural alphabet [1] , i.e. an ensemble of elementary prototype conformations able to describe the whole diversity of protein structures.
- Greedy algorithm HMM-SA letters are assembled by an enhanced greedy algorithm described in [2].
-
Coarse grained force field The
OPEP potential helps us to limit the roughness of the
peptides energetic landscape, by simplifying side chains
representation by a single bead. OPEP v3 parameters are
optimized by a genetic algorithm procedure using a large
ensemble of protein decoys [3]. OPEP
is the objective function that drive the greedy
algorithm during the rebuilding process.
OPEP (Optimized Potential for Efficient structure Prediction) version 3 is expressed as a sum of local, nonbonded and hydrogen-bond (H-bond) terms:
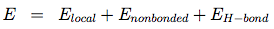
The local potentials are expressed by:
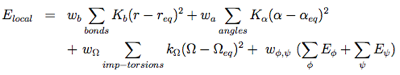
The term Elocal contains force constants associated with changes in bond lengths and bond angles of all particles as well as force constants related to changes in improper torsions of the side-chains and the peptide bonds.
The nonbonded potentials are expressed by:
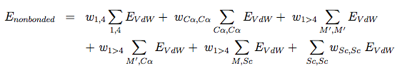
with 1, 4 the 1-4 interactions along each torsional degree of freedom, M ′ the N, C’, O and H main chain atoms, and Sc the side-chain. As seen, we separate short-range from long-range (j > i+4) interactions, and the C alpha atom from the other main chain atoms. For more details on the EVdW potential, see ref [6,5].
The hydrogen-bonding potential (EH−bond) consists of two-body (EHB1) and four-body (EHB2) terms. Two-body H-bonds are defined by:
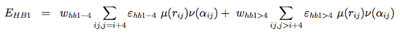
Four-body effects, which represent cooperative energies between hydrogen bonds ij and kl, are defined by:
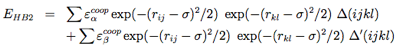
-
Disulfide bonds and residue proximities: The sOPEP force field has been adapted for SSBonds and residue proximities by using a asymetric nonbonded contribution represented below (black: original sOPEP formulation, red: new formulation):
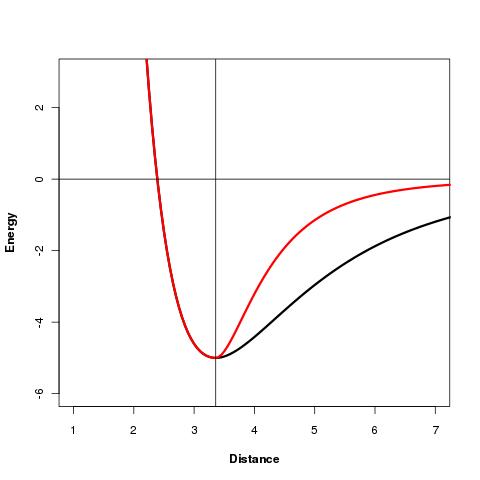
Validation
Validation tests have been performed on two differents
peptides set:
- PepLook cyclic peptide set This test set includes 34 peptides containing disulfide bonds characterized by NMR spectroscopy. [5]. On this test set, PEP-FOLD average RMSd for the best model is of 2.75 A.
- PepStr set This test set includes 42 linear bioactive peptides free of any disulfide bridge characterized by NMR spectroscopy in both aqueous and non-aqueous solutions [4].
-
PEP-FOLD set To complete
PepStr set, we designed our own test set consituted of
two subsets of PDB structures solved in aqueous
solution, selected for their sizes and topology
diversity.
- Short peptides PDB codes (10 targets from 10 to 23 aa) : 1dep, 1k43, 1le1, 1le3, 1pei, 1uao, 1wbr, 1wz4, 2evq and the beta hairpin fragment of 2gb1.
- Long peptides PDB codes (14 targets from 27 to 49 aa) : 1abz, 1aie, 1bbl, 1bdd, 1e0l, 1e0n, 1f4i, 1fsd, 1i6c, 1kjk, 1psv, 1vii, 1vpu and 2p81.
-
PEP-FOLD results on a test set of peptides less than 25 amino acids
(January 2012)
sOPEP: Lowest energy model; Best: Lowest RMSd model (compared to experimental structure); FS: Full Structure RMSd; RC: Rigid Core RMSdsOPEP Best FS RC FS RC 1a13 2.6 2.4 2.4 2.3 1b03 3 3 1.5 1.5 1dep 3.4 3.4 1.5 1.5 1du1 5.2 5.2 3.8 3.8 1e0q 5 5 1.7 1.7 1egs 2.4 2.4 1.1 1.1 1gjf 2.5 2.5 1.4 1.4 1in3 2.6 1.6 2.1 1.7 1k43 2.1 1.4 1 0.8 1l2y 2.5 2.5 2.2 1.9 1l3q 4.4 4.4 2.5 2.5 1lcx 3 2.3 2.4 1.9 1le1 0.4 0.5 0.4 0.4 1le3 1.7 1.7 0.9 0.9 1niz 1.8 1.8 1.1 1.1 1nkf 4.7 4.7 2.1 2.1 1pef 0.7 0.7 0.7 0.7 1pei 2.5 2.5 1 1 1pgbF 1 1 0.8 0.8 1rpv 1.5 0.8 1.1 0.7 1uao 3.5 2.9 0.9 0.7 1wbr 4.8 1.1 3.5 1.2 2bta 4 4 3.1 3.1 2evq 0.4 0.4 0.3 0.3 mean 2.8 2.5 1.7 1.5
Statistics
PEP-FOLD 2013 statistics per country (27898 runs between 2013-01-01 and 2013-12-31)
<Back to top>History
-
2012, Feb 11 - Small adjustments for better SS-bond results.
- 2011, nov 17 - User SS-bond specification available online.
- 2011, sep 17 - Restricted opening of PEP-FOLD improved version, including improved local structure prediction and clustering.
- 2009, sep 17 - Bugfix: in some cases, secondary structure prediction constraints were not efficient.
Known problems and answersThere is presently very few feedback reporting problems with PEP-FOLD. Most are related to Java and browser dependent behavior.
PEP-FOLD has been tested successfully using various OS / browser combinations, including:
Firefox 11 and Chrome 18 under Linux,
Internet Explorer 9, Firefox 3.6 and 11, Chrome 18 under Windows 7, Firefox 7 and Internet Explorer 8 under Windows XP,
Safari 5.1.(2,3), Firefox 11 and Chrome 18 under MacOS X (lion, snow leopard).-
Jmol and OpenAstex applet/viewer will not launch properly In most cases, it is related to (i) security issue related to Java : check for messages asking for permission to launch the applet (ii) Java virtual machine does not behave properly: presently, please prefer official sun/oracle JRE or JDK to other implementations such as openJDK.Results will not display on PEP-FOLD termination - Mobyle upload button will not work. This has been observed in early version of PEP-FOLD 2011, and seems to have been solved. If you encounter such behavior, just perform a full refresh of the result page (press Ctrl R or press Enter in the url specification field of the browser).References[1]
A hidden markov model derived structural alphabet for proteins.
J Mol Biol. 2004 Jun 4;339(3):591-605.[2]
A fast and accurate method for large-scale de novo peptide structure prediction.
J Comput Chem. 2010 Mar;31(4):726-38.[3]
A coarse-grained protein force field for folding and structure prediction.
Proteins. 2007 Nov 1;69(2):394-408.
[4]
PEPstr: a de novo method for tertiary structure prediction of small bioactive peptides.
Protein Pept Lett. 2007;14(7):626-31.
[5]
APOLLO: a quality assessment service for single and multiple protein models.
Bioinformatics. 2011 27(12):1715-6.
[6]<Back to top>
In silico predictions of 3D structures of linear and cyclic peptides with natural and non-proteinogenic residues.
J. Pept. Sci. 2011; epub ahead of print.
Last-Update: 2008/12/18