MTiOpenScreen
A service to dock small compounds.
A service to dock small compounds.
Open screening endeavors play and will play key role in order to facilitate the identification of new bioactive compounds for drug discovery and chemical biology purposes. Such open access tools are critical to advance the success of drug discovery projects in particular for academic groups. This page describes how to use and run MTiOpenScreen web server proposing two services: MTiOpenScreen and MTiAutoDock dedicated to small molecule docking and chemical library virtual screening. The services are hosted on the Mobyle Portal. For specific requirements or reporting possible bugs, please write to us.
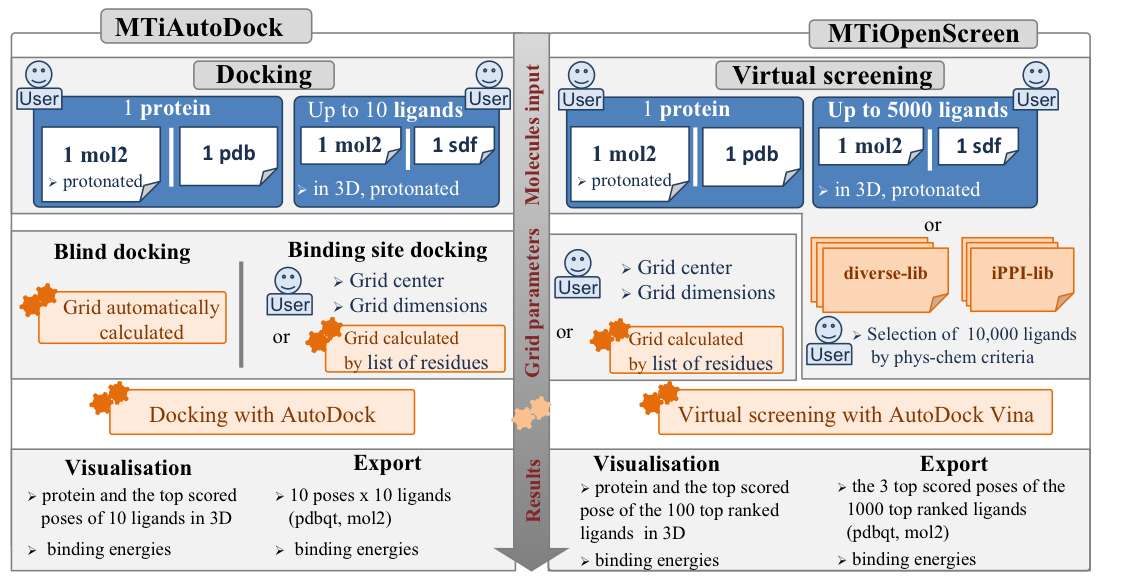
The Lamarckian genetic algorithm (LGA) [1] as implemented in AutoDock 4.2.6 is used to generate orientations/conformations of the compound. Ten docking runs are performed, with an initial population of 150 random individuals and a maximum number of 2,500,000 energy evaluations. The automatically generated grid envelops the entire protein structure.
The Lamarckian genetic algorithm (LGA) [1] is used to generate orientations/conformations of the compound. Ten docking runs were performed, with an initial population of 150 random individuals and a maximum number of 2,500,000 energy evaluations. The grid dimensions and center should be provided by the user or can be automatically calculated based on the list of protein residues of the binding site provided by the user.
AutoDock Vina [2] docking employs a gradient-based conformational search approach and defines the search space by a grid box defined by the box center coordinates and its dimensions of x, y and z. In AutoDock Vina the grid resolution is internally assigned to 1Å. We use number of binding modes of 10 and exhaustiveness of 8. The grid dimensions and center should be provided by the user or can be automatically calculated based on the list of protein residues of the binding site provided by the user. The scoring of the generated docking poses and ranking of the ligands is based on the Vina empirical scoring function approximating the binding affinity in kcal/mol.
To provide valuable starting points for open virtual screening, we provide five electronic drug-like chemical libraries: a diverse chemical compound collection (Diverse-lib) and a focused chemical compound collection (iPPI-lib) to target protein-protein interactions (PPI), a collection of purchasable approved drugs (Drugs-lib), a food constituent compound collection (FOOD-lib) and a natural product compound collection (NP-lib). For the Diverse-lib preparation, we downloaded 12 chemical libraries from PubChem BioAssay Database [3] assembling thus 3,574,650 molecules (names provided in pubchem SID). After removing the redundant molecules, we employed an in-house developed “soft” drug-like filter using the FAFDrugs3 web-server [4] to remove molecules with undesired physicochemical properties (for the drug-likeness parameters see here). Toxic and PAINS (Pan Assay Interference Compounds) groups as defined in FAFDrugs3 were removed (for the toxicophores decided to be removed see here). To ensure chemical diversity, the filtered drug-like 384,372 molecules were then clustered using the Cluster Molecule Protocol (Accelrys Pipeline Pilot v8.5) with the FCFP-4 fingerprint using a maximum distance of Tanimoto of 0.3 in the clusters. Finally, we generated 3D conformations of 99,288 diverse drug-like PubChem molecules, which constitute our predefined diverse compound collection Diverse-lib.
The second provided chemical collection focused to target PPI, iPPI-lib, was prepared starting from the drug-like compound collection containing 384,372 molecules. We then employed PPI-HitProfiler [5] to select PPI-friendly compounds. The program PPI-HitProfiler is based on a machine learning model that was previously trained on 66 chemically diverse low MW inhibitors of PPI and validated using experimental results of 500,000 compounds screened on four PPI targets. The remained 204,728 molecules were then clustered using the Cluster Molecule Protocol (Accelrys Pipeline Pilot v8.5) with the FCFP-4 fingerprint using a maximum distance of Tanimoto of 0.3 in the clusters. Finally, we generated 3D conformations of 51,232 drug-like molecules likelihood to inhibit PPI.
The 3D structures of the two previous collections, the diverse one Diverse-lib and the PPI-focused one iPPI-lib, were generated using the freely available web-server Frog2 [6]. The procedure was launched keeping a maximum of 1 stereoisomer per compound without generating multiple ring conformations. The molecules were finally protonated at pH 7 using the major macrospecies option of the ChemAxon calculator plugins.
The Drugs-lib, FOOD-lib and NP-lib were built using the same protocol [7] that includes the use of FAF-Drugs4 [8] physico-chemical and toxicophore filtering, a visual inspection step to remove compounds not suitable for docking and the assessment of their purchasability according to the ZINC15 database [9]. The Drugs-lib was constructed by using as a starting point four databases of approved drugs, the "drug" subset of the ChEMBL database [10], the "approved" subset of DrugBank [11] version 5.0.10, the DrugCentral online compendium [12] and the "approved" SuperDrug2 database version 2.0 [13]. A total of 21276 compounds were initially included in the Drugs-lib construction process, but only 7173 stereoisomers corresponding to 4574 single isomer drugs were kept after removal of the duplicates and the filtering protocol mentioned in the method section. The 26941 food constituents of the FooDB with available smiles strings were used as input for the FOOD-lib generation. After removal of the duplicates and the filtering steps, the FOOD-lib included 10997 stereoisomers corresponding to 3015 single isomer food constituents. A purchasable diverse library of natural products of 1237 compounds [14] was processed to construct the NP-lib that gathers, after the filtering steps, 1228 stereoisomers corresponding to 653 single isomer natural products.
The “New Job” pages of MTiAutoDock and MTiOpenScreen guide the user for the job submission. New jobs can be easily submitted by providing the protein receptor structure in PDB or MOL2 (protonated protein structure is required for MOL2) along with binding site definition, and ligands to be uploaded or drug-like compounds provided in the MTiOpenScreen webserver to be selected.
The user can chose to proceed docking of up to 10 ligands for a protein receptor provided by the user via AutoDock; virtual screening of up to 10,000 compounds on one protein conformation using AutoDock Vina.
For protein receptors, if they are provided in PDB, the structure are cleaned and preprocessed automatically: all HETATMs are removed and hydrogens are added to the structure using MGLTools. In most cases, these procedures will produce a clean receptor structure. Otherwise, users can prepare the protein structure manually and upload it in MOL2 format. If the protein is uploaded in MOL2, the structure will be used as-is without any modification. If MOL2 is used, please remove all the solvent molecules and add all hydrogens. If the protein of a bound complexe is used as the receptor, please do remember to remove the native ligand structure from the complex before uploading.
The user should define the binding site (except for the blind docking) by providing the center and the grid dimensions, or a list of residues defining the binding site.
For ligands, a unique conformation should be given for up to 10 small organic molecules with added all hydrogen atoms in a SDF or MOL2 file. For correct docking results the number of ligand atoms should not exceed 300 atoms. Please, make sure that all the atom types and the bond types of the molecules are correct.
User has the possibility to select one of the following options:
For protein receptors, if they are provided in PDB, the structure are cleaned and preprocessed automatically: all HETATMs are removed and hydrogens are added to the structure using MGLTools. In most cases, these procedures will produce a clean receptor structure. Otherwise, users can prepare the protein structure manually and upload it in MOL2 format. If the protein is uploaded in MOL2, the structure will be used as-is without any modification. If MOL2 is used, please remove all the solvent molecules and add all hydrogens. If the protein of a bound complexe is used as the receptor, please do remember to remove the native ligand structure from the complex before uploading.
The user should define the binding site (except for the blind docking) by providing the center and the grid dimensions, or a list of residues defining the binding site.

Upon successful submission of a new job, a unique token will be given to identify the job. Use this token to check the status of the job and retrieve the results.
When the job finishes, the results will be parsed and visualized. The receptor and docked compounds will be visualized in 3D:
The virtual screening results are also available for downloading. The user can download:
Please, note that bioactive compounds are rarely ranked in the top 1 % of the virtually screened compound library. Thus, the visualized here top ranked 100 ligands using MTiOpenScreen are only an illustration of the docking results and the binding site. You may need to proceed a thorough analysis of a larger number of ligands among the 1500 top ranked compounds provided to download by using standalone programs like PyMOL or AutoDockTools for a final selection of the best potential compound candidates.
The data associated with any job will be kept for only a limited time period. All jobs terminated due to any error will be deleted within 24 hours and all successfully finished jobs will be kept for only 30 days.
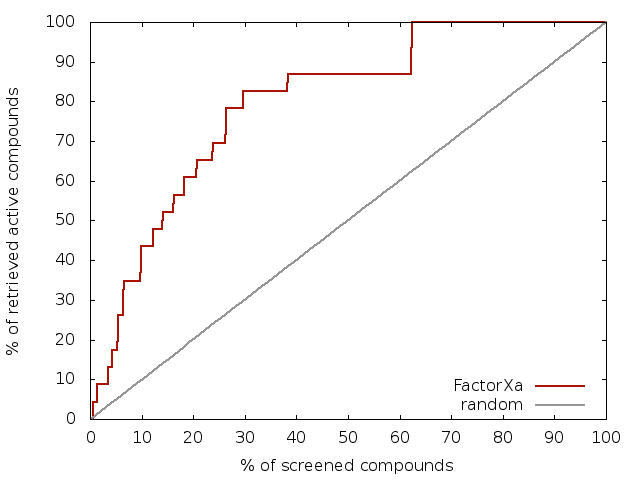
MTiOpenScreen has been rigorously benchmarked for the performance of our implementation of AutoDock and Vina software. Docking accuracy has been validated on 27 crystal structures of protein-drug complexes taken from the DrugPort database of EMBL-EBI. We selected several classes of important protein targets (like nuclear receptors, GPCR, kinases, serine proteases, PPI among others) with available high quality crystal structures that have been manually verified for the electronic density quality using VHELIB. The ability to discriminate small-molecule binder from non-binder compounds has been assessed on two enzymes (the catalytic domains of the RTK VEGFR2 and the serine protease Coagulation Factor Xa) and on the PPI target Bcl-xl using up to 40 diverse active compounds and ~1000 diverse decoys per protein target. An example is shown for the enrichment obtained for the catalytic domain of the serine protease Coagulation Factor Xa after virtual screening with MTiOpenScreen.
AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility.
J Comput Chem. 2009, 30(16):2785-91.
AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading.
J Comput Chem. 2010, 31(2):455-61.
PubChem BioAssay: 2014 update.
Nucleic Acids Res. 2014, 42(Database issue):D1075-82.
The FAF-Drugs2 server: a multistep engine to prepare electronic chemical compound collections.
Bioinformatics. 2011, 27(14):2018-20.
Designing focused chemical libraries enriched in protein-protein interaction inhibitors using machine-learning methods.
PLoS Comput Biol. 2010, 6(3):e1000695.
Frog2: Efficient 3D conformation ensemble generator for small compounds.
Nucleic Acids Res. 2010 38(Web Server issue):W622-7.
Online structure-based screening of purchasable approved drugs and natural compounds: retrospective examples of drug repositioning on cancer targets.
Publication submitted.
FAF-Drugs4: free ADME-tox filtering computations for chemical biology and early stages drug discovery.
Bioinformatics. 2017, 33(22):3658-60.
ZINC 15--Ligand Discovery for Everyone.
J Chem Inf Model. 2015, 55(11):2324-37.
ChEMBL: a large-scale bioactivity database for drug discovery.
Nucleic Acids Res. 2012, 40(Database issue):D1100-7.
DrugBank 5.0: a major update to the DrugBank database for 2018.
Nucleic Acids Res. 2018, 46(D1):D1074-82.
DrugCentral: online drug compendium.
Nucleic Acids Res. 2017, 45(D1):D932-9.
SuperDRUG2: a one stop resource for approved/marketed drugs.
Nucleic Acids Res. 2018, 46(D1):D1137-43.
Analysing and Navigating Natural Products Space for Generating Small, Diverse, But Representative Chemical Libraries.
Biotechnol J. 2018, 13(1).