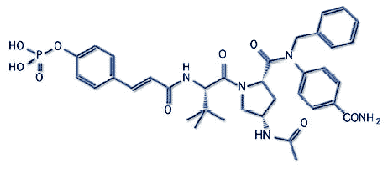
| c1(nn(c(c1Br)C)CC(Nc1ncccc1C1CCC[NH+]1C)=O)[NH](O)=O | ==> | #1:
c1(nn(c(c1Br)C)CC(Nc1ncccc1[C@H]1CCC[N@H+]1C)=O)[NH](O)=O #2: c1(nn(c(c1Br)C)CC(Nc1ncccc1[C@H]1CCC[N@@H+]1C)=O)[NH](O)=O #3: c1(nn(c(c1Br)C)CC(Nc1ncccc1[C@@H]1CCC[N@H+]1C)=O)[NH](O)=O #4: c1(nn(c(c1Br)C)CC(Nc1ncccc1[C@@H]1CCC[N@@H+]1C)=O)[NH](O)=O |
1: 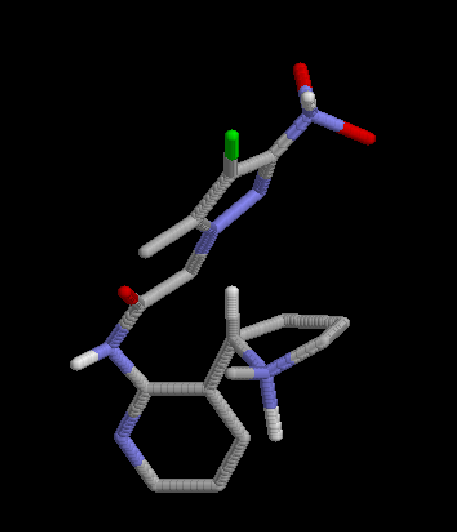 |
2: 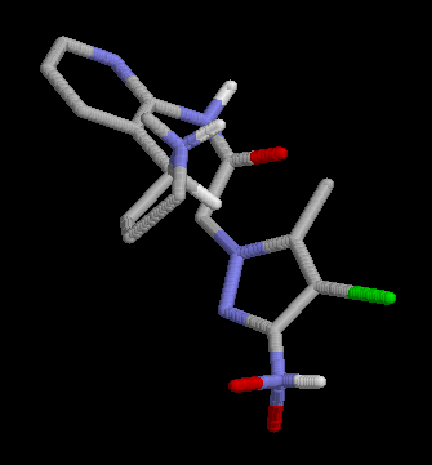 |
3: 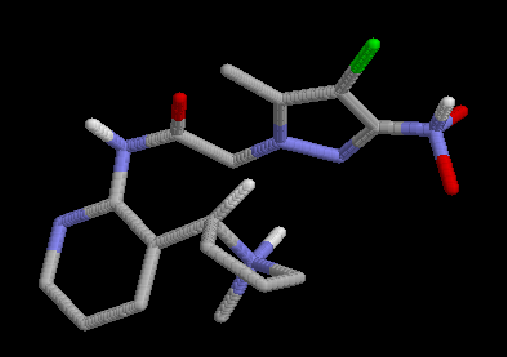 |
4: 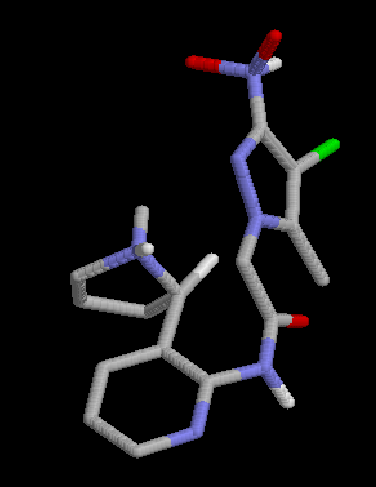 |
1: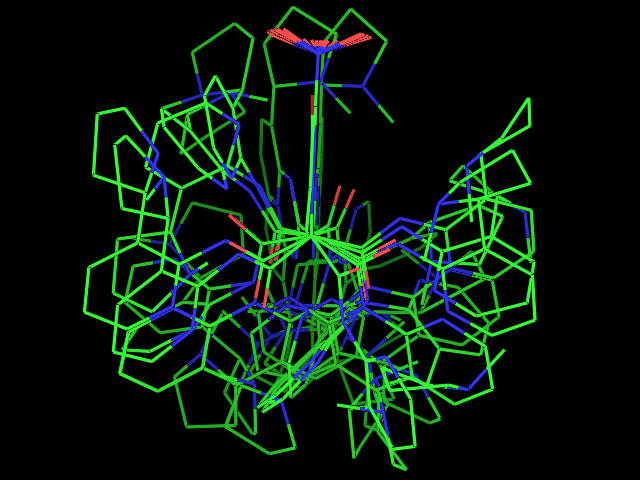 |
2: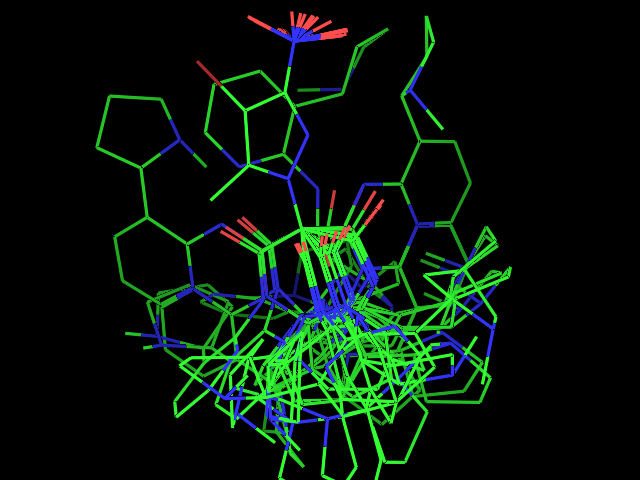 |
3: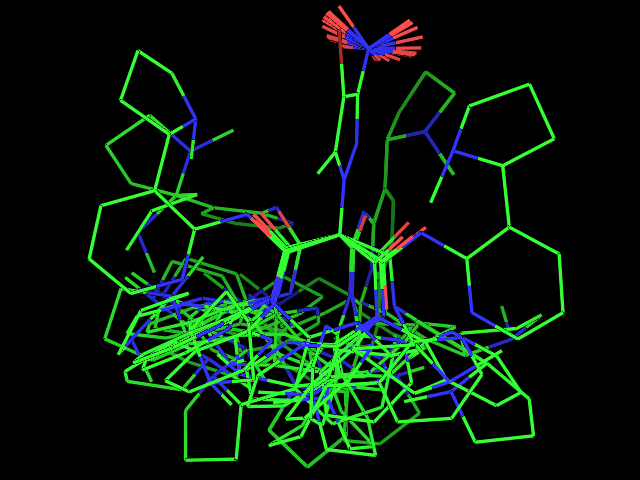 |
4: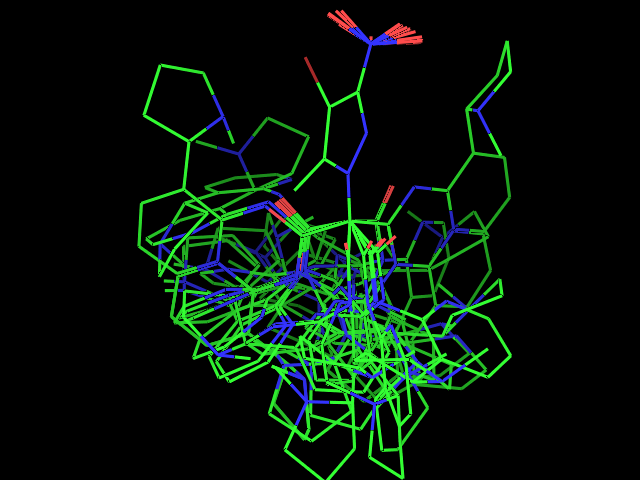 |
1. History
2. Features
3. Limitations
4. Usage
5. Time considerations
6. Examples, sample tests
7. Concepts
8. Validation
9. Availability (news since 2009 January)
10. Citations
History:
- 2006: Frog1: analysis of ambiguous compounds, energy assessment, towards multi conformation (T. Bohme Leite). Intensive checks of isomer detection (chirality, ZE conformations, axial/equatorial). Check of 3D assembly (mono conformation). Hydrogen positioning.
- late 2006: energy assessment of conformations (based on an implementation of Merck Molecular Force Field (MMFF)).
- February 2007: small bug fixes (Force Field parameters).
- January 2009: redesign of conformation generation, better ring
hydrogen management.
- February 2009: redesign of Frog conformational sampler to enhance
diversity.
- August 2009: interface with DG-AMMOS and AMMOS to solve on the fly ring generation and energy minimization.
- November 2009: Frog2.0b released
- February 2010: Frog2.1 released (better diversity, enhanced speed).
- March 2010: Frog2.12 released
- better management of possibly incorrect conformation by automatic trigger of fast minimization.
- better information about some unmanaged chiral center(s) located in bridged rings, thanks to Eizo AB.
- April 2010: Frog2.13
- better management of 3D stereoisomery
- January 2011: Frog2.14
- bug fix: better management of 3D stereoisomery at ring/appendix junction, thanks to J.-P. Ebejer.
- August 2011: Frog2.14 source code released under the GPL license.
- See the Availability section.
- Solve isomer ambiguities occurring in compounds expressed
using the 1D SMILES[1] or 2D SDF [2] formats, used by most academic
or commercial compound collections. Frog will process the input data, identify chiral centers and produce a list of unambiguous smiles, each smiles corersponding to one unambiguous isomer . Frog will also consider axial / equatorial conformations for cycles when relevant. Note however that Frog does not consider some stereo centers in some bridged cyclic systems since it presently only considers one conformation per ring. - Generate from SMILES, SDF or Mol2 [3] 3D coordinates for the
compounds. It
is possible to ask for multi conformations per isomer. Multi
conformations are often of great help in the process of in silico
compound screening. While relying on a ring library, Frog2 is able to
generate on the fly ring conformations thanks to DG-AMMOS[4]
- Generate from 3D SDF or 3D Mol2 ensemble of conformations from a
starting conformation.
- Minimize conformations generated using AMMOS[5].
2: Arthur Dalby et al., J. Chem. Inf. Comput. Sci, 1992, 32, 244-255
3: Tripos Mol2 File Format
4: Lagorce D, Pencheva T, Villoutreix BO, Miteva MA. DG-AMMOS: a new tool to generate 3d conformation of small molecules using distance geometry and automated molecular mechanics optimization for in silico screening. BMC Chem Biol., 2009, 9, 6.
5: Pencheva T, Lagorce D, Pajeva I, Villoutreix BO, Miteva MA. AMMOS: Automated Molecular Mechanics Optimization tool for in silico Screening. BMC Bioinformatics. 2008, 9, 438.
Limitations:
- Frog build compounds involving atom types commonly accepted as
non-toxic for drugs (i.e. C, N, O, P, S, H, etc).
Ions present in salts can however be removed before processing by the facility
accessible from the form.
- Frog multiconformation generation has an enhanced sampling algorithm. However, for compounds having large number of degrees of freedom the combinatorial search is biased to keep reasonable computational time.
- Frog energy scoring is based on MMFF [6]. Although validation has been performed, some particular molecular arrangements can fall out of our present implementation.
- Protonation: although Frog will generate hydrogen coordinates, Frog manages protonation in a standard way (based on openbabel). It is best to re-protonate the conformers outside Frog, depending on the pH conditions.
- Number of compounds per run: Frog2 is presently limited to 5000 compounds per run. Larger collections are possible on request to the authors.
- Speed: Although Frog2 is much faster than Frog1, calculations remain costly for large sets. See the examples section.
- Erroneous conformations: Frog performance relies on the quality of the input. Most errors come from poor input specification. See the input section for more information. Note: Frog will sometimes fail depending on the input format of a compound. In such case, resubmitting using another format may solve this (under investigation on date of December 26th, 2009).
- Bridged rings can exhibit asymmetry in some cases. Presently, since Frog only considers one conformation per ring some stereoisomers may be discarded. Specified stereoisomery on such stereocenters involving bridged cyclic systems will not be taken into account. Only one conformation (of unspecified stereoisomery) of the cyclic system is currently considered.
- Large compounds involving large
bridged rings (over 30 atoms) that might be flexible are
presently not in Frog scope.
6: Halgren T. A.; Merck Molecular Force Field: I. Basis, Form, Scope, Parameterization, and Performance of MMFF94 (490-519), II. MMFF94 van des Waals and Electrostatic Parameters for Intermolecular Interactions (520-552), III. Molecular Geometries and Vibrational Frequencies for MMFF94 (553-586), IV. Conformational Energies and Geometries for MMFF94 (587-615), V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules (616-641). J. Comp. Chem., 1996, Vol.17, Nos. 5 & 6
Usage:
- Input type: by default, Frog will consider
input as 2D input. Thus 3D conformations will be generated ffrom
scratch. Use the 3D input type to generate multiconformation of the
input data.
- Input format: smiles , sdf and mol2 formats are accepted for the compounds. In order not to overload the server, requests are limited to 5000 compounds. It is possible to both upload a file and paste data. However, the two body of data MUST be on the same format (i.e. both are smiles, both are sdf or both are mol2).
For scratch generation, prefer not specifying the hydrogens in the input.
- Output: 3D files can be returned using the
PDB, SDF or mol2
formats. 1D information is returned using the SMILES format.
Unambiguate results are always smiles. Identifiers of the returned
conformations are on the form: inputIdentifier_#isomer_#conformation.
3D conformations with energies more than MaxE are returned in a
separate (_BadEnergy) file.
- Disambiguate: Attempt to indentify atoms for which isomery is not specified, and generate a set of unambiguous smiles (i.e. one per isomer identified). Too large combinatorials are truncated to a maximum of 8 isomers.
- stage2MC: Frog multiconformation
generation is based on a two stage Monte-Carlo. The first in the the
space of representative dihedral values (few values per dihedral). The
second is a refinement stage using smaller rotation values, in order to
solve, for some conformations, clashes resulting from the coarse
grained sampling of stage one. You can switch off the second stage
Monte-Carlo, so as to gain in speed. However, this is sometimes at the
cost of some loss in the conformational diversity of the returned
conformation ensemble.
- Minimize: The conformers generated will
undergo further energy minimization using AMMOS. Note: this can result in side
effects for conformational diversity since several conformations could
converge after minimization, depending on the context. Also note that this has a
computational cost that is not marginal for large sets. Since April 2010, Frog2 embbeds a new
default "Auto" strategy to
automatically trigger the minimization of compounds of high energy.
This will affect the speed of the conformation generator since
minimisation is costly. You can switch this option to "No" to gain speed.
- Processing: the user can choose among "Unambiguate", "Single" and "Multi".
- "Single" will generate one conformation per unambiguous isomer of each compound.
- "Multi" will generate upto #conf conformations per isomer for each compound.
- #confs: Maximal number of conformations returned per isomer. The actual number depends on acceptance of the conformations due to the EMax threshold.
- over: the maximal numer of conformer can
be expressed on a per isomer basis (at maximum #confs per stereoisomer,
thus at maximum #confs x n stereoisomer on output) or overall (over all
stereoisomers, a maximum of #confs will be returned (thus Frog
only attempts to generate #confs / n are generated per stereoisomer). Note: the actual returned number of
conformations returned by Frog might differ slighty above the overall
number of conformers requested if several stereo-isomer are present,
since Frog will attempt to generate the same number of conformations
per isomer.
- E Window: Maximal Energy difference to
lowest
energy conformer to flush 3D
conformations. Conformations with scores
more than this value will be discarderd. This energy window is in terms
of Frog2 internal energy (not all contributions are considered, only
some Vand der Waals terms are calculated: contribution from rigid parts
such as rings are not calculated). Since no energy optimisation is
performed, a value of 50 kcal/mole performs usually well.
- E max: Maximal energy value to accept
conformer as plausible. Compounds for which the lowest energy is above
this value will be placed in the high energy compound result file. Note: since February 18th, 2010, AMMOS
energies are used to check this. Since March 05th, 2010, these energies
are used to automatically trigger some minimization steps to reduce at
maximum the number of bad energy conformers.
- RMSd: RMSd value for 3D clustering (not two
conformers should be closer than this value). Note: Ring symmetries are presently not
fuly considered. This can result in apparently identical conformers.
- The returned log will give information about the treatment of
each compound: smiles, axial-equatorial conformer string if relevant (A
for axial, E for equatorial, the string matches the smiles' heavy
atoms). Errors can occur (see limitations).
Using "Quick3D" or "Single", the first conformation not presenting strong steric clashes will be returned.This is intended to provide rapidly a correct 3D geometry of the compound
for one or all its isomers.
Only using "Multi", conformations of low energy are returned. Be aware that this is under current optimisation on two directions: (i) relevance of the lowest energy
conformations (ii) computational speed. At present too large compounds still require important computational time. This is under investigation.
Note on formats:
- Smiles files should be on the form of one smiles per line, such as:
O=C(CCCCCCCCCC[C@H]1[C@@H]2[C@@H](c3c(C1)cc(cc3)O)CC[C@@]1([C@@H](CC[C@@H]21)O)C)[N@](C)CCCC another_compound_identifier
O=C1[C@@H]2[C@H](N[C@H](N1)N)[N@](CCC(CO)CO)[CH]N2 TKinh5_penciclovir_1KI3pdb
- The mol2 format is described here.
- The sdf format should be on the form (Note the $$$$ line):

Time considerations:
Frog2 engine is, in our assessment, much faster than Frog1'. On the astex test set (83 compounds) Frog2 (off-server) required 11 minutes.
Using comparable parameters, Frog1 generated the same collection in 103 minutes. The server calculation times are however usually slower than this, since several additional
operations such as image generation are performed, and since the calculations are performed on a cluster on which the cpu load is variable.
Examples:
- Smiles input disambiguation
CC(=C(C)C(O)C)F
Select Unambiguate.
Resulting smiles are:
CC(=C(/C)[C@H](O)C)/F
CC(=C(/C)[C@H](O)C)\F
CC(=C(/C)[C@@H](O)C)/F
A more complex sample test (19 smiles) can be accessed here.
The unambiguation results (38 smiles) can be accessed here.
- 3D generation from scratch
The results of the multi generation (10 conformations at max per isomer) can be accessed here. (mol2 format, 275 conformations) (note: for some compounds, less than 10 conformations of low energy were identified).
Some 3D conformations generated using Frog2.0b (multiconformations) - starting from these smiles representation, asking for no disambiguation, energy window of 25 kCal/mole, 50 conformers - , for 103 compounds of the astex collection for which experimental data is available here can be accessed here (2637 conformers).
- 3D generation from 3D input
- Examples of conformational sampling
Left: experimental conformation Right: diversity of 50 conformations generated using Frog2, from scratch (removing 3D information prior to 3D generation) |
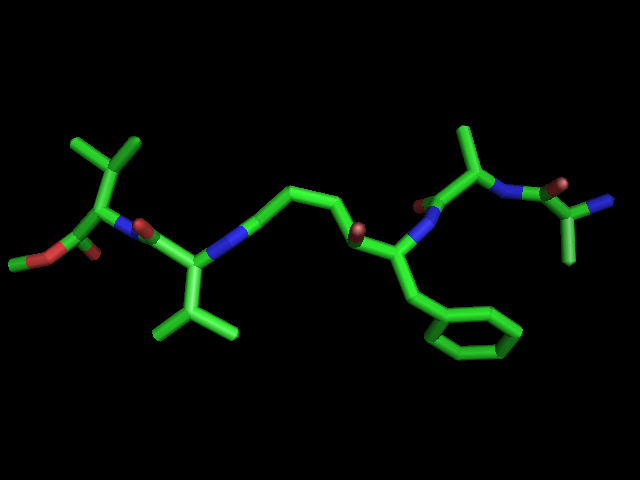 |
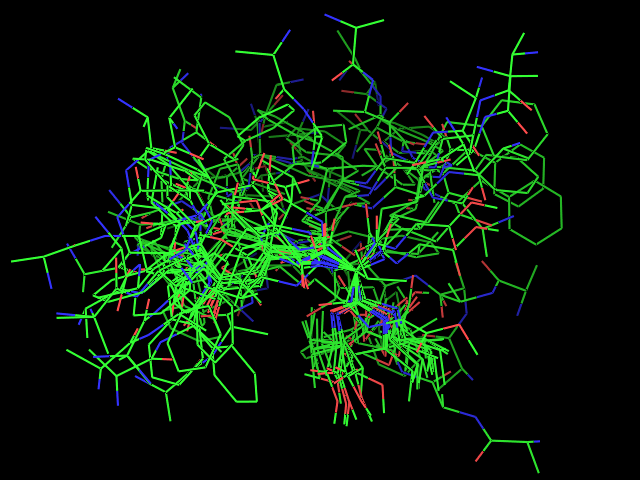 |
Left: experimental conformation. Right: diversity of 50 conformations generated using Frog2, from scratch. |
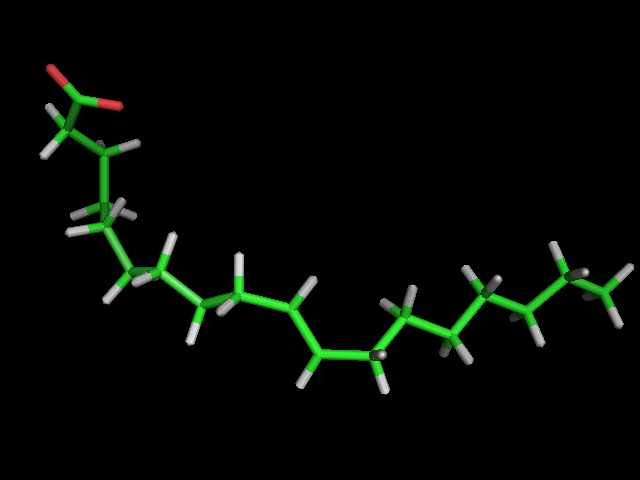 |
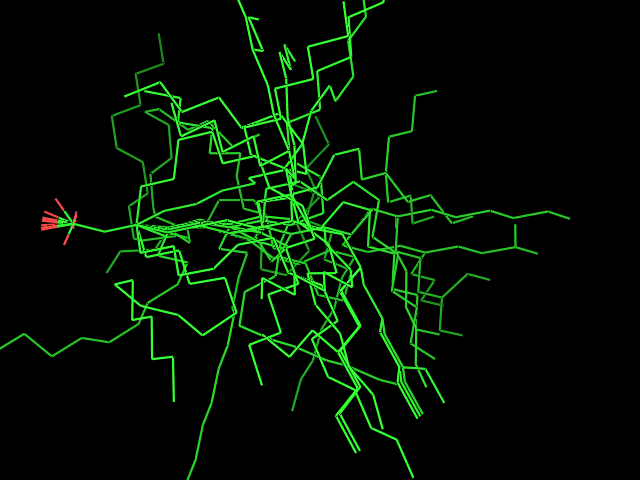 |
Older tests:
Random test upon 992 compounds from Specs, Chembridge and Ambinter,
using as values of energetic treshold of 100.0,
number of Monte Carlo steps of 100, number of conformations of 10.
The input smiles are here.
The unambiguated smiles (1238) are here.
The mol2 output (12668 conformers) is here.
The log file is here.
Compounds not processed (might not be ADME/Tox compliant) here.
Note: the number of 12668 (i.e. more than 1238 x 10) is due to
componds for which axial equatorial conformers have been considered.
See for instance compound Chembridge-6439335, 2 smiles to describe the
isomers, but 4 conformers considered due to axial/equatorial
conformations.
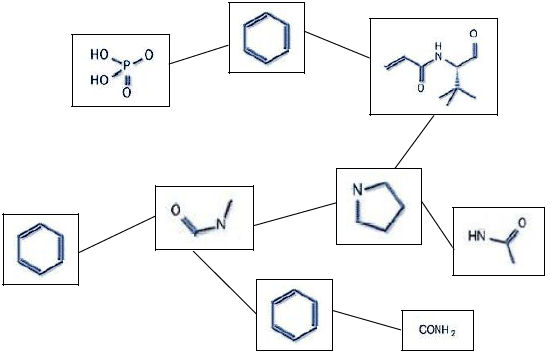
- Graph approach to compound 3D generation. Nodes types are cycles,
linkers, appendices. (see image below)
- Rings correspond to simple or complex rings made of several simple rings connected together (sharing atoms).
- Linkers are compound fragments that interconnect cycles.
- Appendices are fragments that are bound to cycles, not linking several cycles.
- Ring flexibility is not addressed by Frog. Cycle
conformations are taken from a library of cycles extracted from
collections of publicly available collection of 3D compounds. Such
strategy has already been described [7]. Frog2
revisits it by adding
the possibility to generate on the fly ring conformations, adding them
to the ring library.
- Flexibility of compounds results from dihedrals of the linkers and the appendices. Covalent geometry flexibility is ignored.
- Ring multi conformation not presently considered, although it is conceivable from the different conformations extracted, stored in the library.
- Multi conformations are obtained by sampling the flexible
dihedral angles of the compounds, sorted according to their MMFF
energy. This has several limitations. The two major are: (i) the non
relevance of MMFF to reproduce the relative orientation of cyles in
some cases. (ii) For large compounds, the combinatorial to explore is
huge. Frog will presently truncate it. In Frog2,
conformations sampling
is enhanced to avoid conformation redundancy based on at least one
dihedral canonical value difference for two conformers. Further
improvement of this strategy is still possible.
7: Sadowski, J.; Gasteiger, J. From Atoms and
Bonds to Three-Dimensional Atomic Coordinates: Automatic Model
Builders. Chem. Rev. 1993, 93, 2567-2581
 |
==> |
 |
Validation tests:
- Chirality detection on the
Asinex library: Selection of 84.812
compounds for which some chirality information was present. Removed
chirality information for the compounds, ask for unambiguation. Check
if original chirality information regenerated. Success for 84792 (over
99%). After manual analysis, the 20 remaining compounds are false
negatives!
- Isomery 3D assembly: random selection of compounds over Asinex, Ambinter, Specs et ChemBridge. 3D generation. Visual inspection of all the isomers: OK.
- Atom types assignment: performed using the MMFF94 validation suite (235 compounds).
- Some 3D conformations generated using Frog, for which
experimental data is available here
can
be accessed here.
- Some results obtained for the astex diverse test set are summarized here
- Larger scale tests for 3D multiconformation:
Availability:
- January 2009: Frog v1.0 is freely available under the terms of the GNU GPL license. You can access the Frog source code here. The authors appreciate if you send an email, so as to identify Frog users, and send news about Frog evolution.
- December 2009: Frog2 will
be made available under the same terms shortly (further packaging
required).
- March 2010: Frog2
available upon request to the authors, for tests, waiting for free
final release.
- August 2011: Frog2
source code available under GPL license on https://github.com/tuffery/Frog2.
Using Frog, please cite:
- Frog: a FRee Online druG 3D conformation generator. Leite TB, Gomes D, Miteva MA, Chomilier J, Villoutreix BO, Tufféry P. Nucleic Acids Res. 2007 Jul;35(Web Server issue):W568-72. Epub 2007 May 7.
- Frog2: Efficient 3D conformation ensemble generator for small compounds. Miteva MA, Guyon F, Tufféry P. Nucleic Acids Res. 2010 Jul;38(Web Server issue):W622-7. Epub 2007 May 7.