DaReUS-Loop
A web server to model loops in homology models
A web server to model loops in homology models
DaReUS-Loop is a web server for the (re-)modeling of loops in homology models. It follows a data-based approach, identifying loop candidates by mining the complete set of experimental structures available in the Protein Data Bank (PDB)[1].
Candidate loops are filtered based on the sequence and then ranked using the local conformation profile and the structural fit. DaReUS-Loop returns ten loop models for each individual loop region.
DaReUS-Loop requires an initial homology model, and can be used for either loop modeling or loop remodeling. For a single loop region, the results are identical. For multiple loop regions, remodeling typically gives more accurate results.
Access the service through the RPBS Mobyle portal:
When using this service, please cite the following reference:
DaReUS-Loop: a web server to model loops in homology models.
Nucleic Acids Res. 2019 July 02.
DaReUS-Loop is used to either do loop modeling or loop remodeling. In both cases, DaReUS-Loop requires an input structure and an input sequence.
For loop modeling, the input structure must be a gapped PDB, whereas the input sequence must contain the full protein sequence.
For loop remodeling, it is the reverse: the input structure must contain a complete initial homology model, whereas the input sequence must be gapped, indicating the loop regions that will be remodeled.
In general, best results are obtained if a (re)modeled loop is flanked by helices or strands. If this is not the case, you might consider to extend the loop region towards the nearest helix or strand.
Typical runs require on the order of one hour, depending on server load.

Indicates whether loop modeling or loop remodeling is to be performed. For a single loop, the results are identical. For multiple loops, loop remodeling is typically more accurate.
It is also possible to do "advanced loop modeling", causing all loops to be modeled completely independently. This is more accurate than standard "modeling mode" (but typically still less accurate than "remodeling mode"). However, each result will contain the coordinates of only one loop, with gaps for all other loops.
DaReUS-Loop requires an initial homology model to (re)model the loops. The input file must be in PDB format. It must contain only the 20 standard amino acids. Homology models can be obtained from e.g. the Swiss-Model server [7] or the ModBase database [8].
DaReUS-Loop specializes in the (re)modeling of loops in homology models. While it is possible to upload experimentally solved structures, the DaReUS-Loop protocol has not been optimized for this. In addition, please note that experimental structures often contain extra atoms (water molecules, co-factors, etc.) which are not accepted by the DaReUS-Loop server and must be manually removed.
In loop remodeling mode, the homology model file can be uploaded directly. In loop modeling mode, the coordinates of the loop regions that are to be modeled should be removed from the file.
In loop modeling mode, this is the full sequence of the protein that is to be modeled. It may contain only standard amino acids.
In loop remodeling mode, the sequence of the loop regions that are to be remodeled should be replaced by gaps. "-" and lower-case amino acid letters are recognized as gaps.
In all cases, the protein structure and the protein sequence must follow exactly the same numbering scheme. For example, if the protein has residue "ALA 16", the 16th letter of the sequence (including gaps) must be "A".
By default, DaReUS-Loop searches the entire Protein Data Bank (PDB) for suitable loop candidates. It is possible to define a PDB code that will be removed from the search, including its close homologs (> 70 % sequence identity). This is useful for benchmarking purposes.
DaReUS-Loop requires an initial homology model to (re)model the loops. The model must already have the correct sequence, i.e. raw template structures are not accepted.
The residue numbering must start from 1.
DaReUS-Loop can model the loops of a single protein chain, that may contain only standard amino acids.
DaReUS-Loop will model missing side chain atoms using oscar-star [5], but all backbone atoms must be present.
The protein sequence may contain only standard amino acids.
The protein structure and the protein sequence must follow exactly the same numbering scheme. For example, if the protein has residue "ALA 16", the 16th letter of the sequence (including gaps) must be "A".
Loops are represented as gaps in the input sequence or structure. Between two gaps, at least four non-gap residues must be present.
The recommended minimum loop length is 4, and the maximum loop length is 30.
DaReUS-Loop cannot model N- or C-termini. It is still recommended to provide full sequences rather than truncated ones, since this leads to better sequence profiles. Otherwise, missing N- or C-termini in sequence or structure are ignored.
The maximum number of loops in the structure is 20.
DaReUS-Loop assumes that, other than the (gapped) loop regions, the initial homology model is of decent quality (TM-score > 0.5). In particular, the flank regions (the four residues adjacent to each gap) must be accurate. It is highly recommended to define gaps such that all flanks are in a helix or sheet region of the homology model.
The demo example is CASP11 target T0762 (atmB transport protein), which has been solved as PDB code 4Q5T. The initial homology model was built using MODELLER [11], using as a template PDB:2V25 (Peb1A Asp/Glu receptor, 19 % sequence identity), except an N-terminal motif which comes from PDB: 2LK9 (BST-2 transmembrane helix). Templates were identified from sequence using HHSearch [9] and optimally aligned using TM-Align [10]. A gapped sequence was defined corresponding to four loop regions where the template was missing, extended until all flanks were on helix/sheet regions of the model.
DaReUS-Loop returns ten models for each loop that is being modeled. On-line interactive visualisation and model selection facilities are offered.
The aim of DaReUS-Loop is to achieve a high accuracy for at least one of those ten loop models. In addition, a confidence score is presented. The better the confidence score, the higher the expected accuracy of the best loop.
DaReUS-Loop does not attempt to predict the best among the ten loop models. Moreover, in case of multiple loop regions, DaReUS-Loop does not attempt to predict which combination of loop models is the best. While such predictions would be very useful, we have not yet found any algorithm, score or potential that can do this successfully. Experimental data, e.g. SAXS data, could in theory be used to filter the combinations.

This section will incrementally provide information about job progression and errors if any. A typical run should produce a report similar to that. Errors related to the input data specified are now also reported in this section.

DaReUS-Loop on-line interactive visualization of the models generated is based on the NGL . Different representations can be selected. A menu makes it possible to select a model among the 10 loop models, or to visualize them together.

This section reports the confidence of loop modeling results. For ever loop, the confidence score and level are indicated. The better the confidence score (smaller values), the higher the expected accuracy of the best loop.
This archive contains all 10 models for all loops in PDB format. It is in the unix tar format compressed using gzip.
Once saved on your computer, enter for instance (unix) tar xzf AllModels.tgz to inflate the archive.
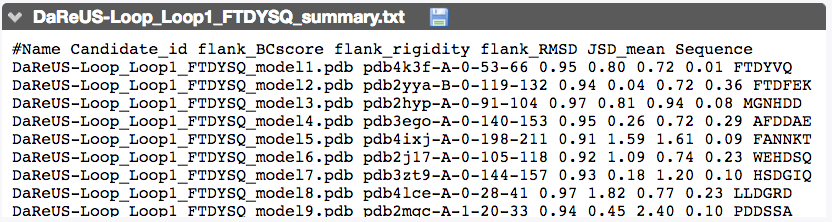
This section reports the detail corresponding to the best candidates identified for every loop. For each candidate, we indicate the PDB entry, the chain, the residue numbers corresponding to the fragment, BCscore, flank rigidity, flank RMSD, Jensen-Shannon divergence and loop sequence.
The example corresponds to the third demonstration mode .
The result page of this demo can be accessed here.
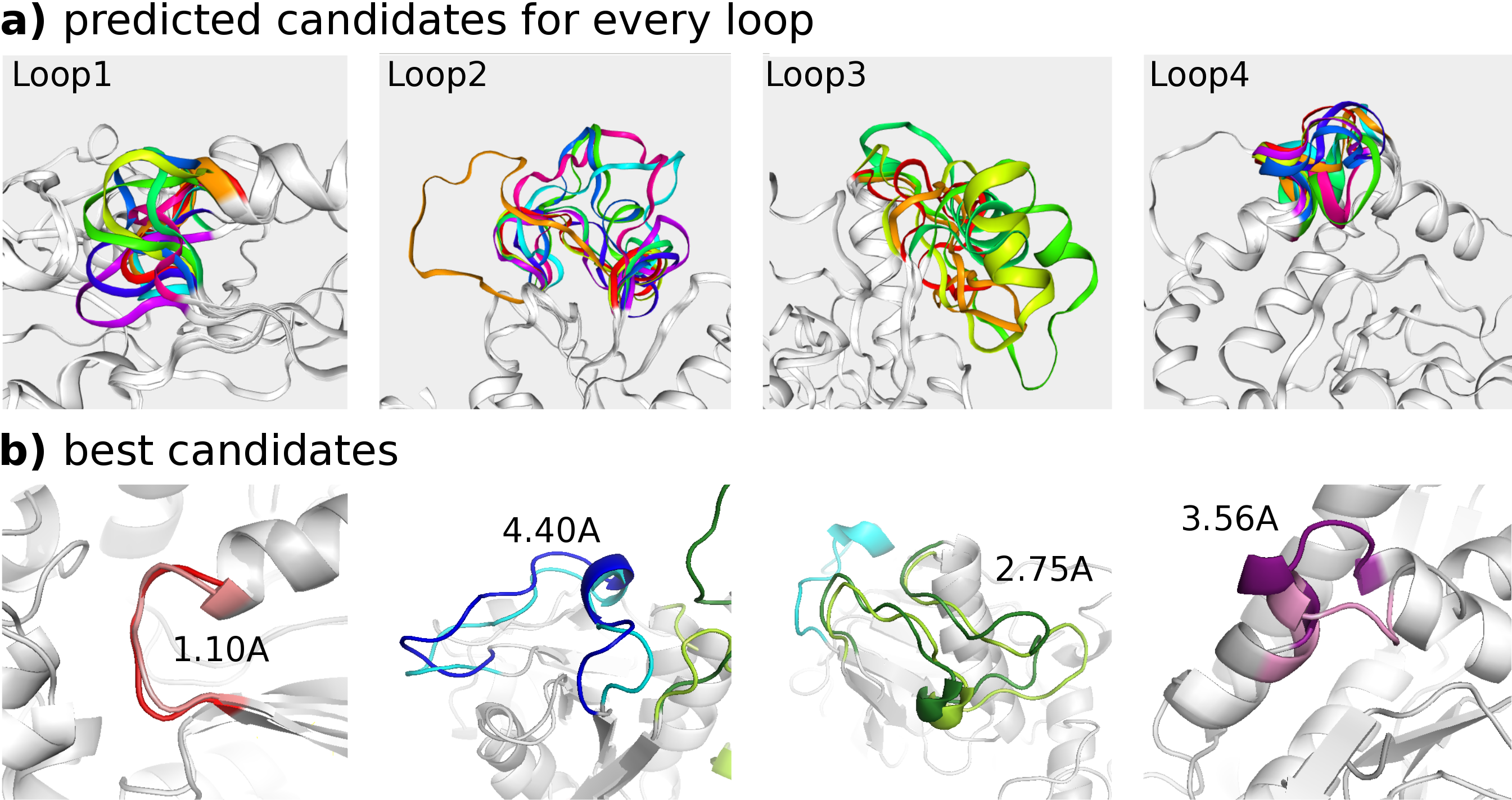
The gapped sequence defines four loops, ranging 66-71, 120-137, 159-184 and 259-265. In the initial homology model, the loop RMSDs are 1.85, 4.46, 18.58 and 5.37 A (after fitting on the flanks). These values reflect the difficulty of loop modeling for this protein. Between the top candidates predicted by the web server, loop candidate #1, #3, #1 and #7 have an improved loop RMSD of 1.10, 4.40, 2.75 and 3.56 A, respectively (after fitting on the flanks). For each loop, the results for (a) all candidates and (b) the best candidate are shown in the figure below.
DaReUS-Loop is based on considering a single loop region at a time (in parallel), keeping the other loops constant. We support three different approaches on how to treat these other loops. Note that this is irrelevant if there is only one loop region.
When tested on a benchmark, we found remodeling mode to be (on average) the most accurate, followed by advanced modeling mode, followed by modeling mode.
DaReUS-Loop is based on the identification of candidate loop structures matching the geometrical conditions imposed by the flank regions. Such fragments are identified by mining the complete collection of protein structures of the PDB, using the previously published protocol protein loops [2], i.e. an approach searching for candidate fragments matching satisfactory geometrical contraints on the loop flanks [3][4]. Candidates with a good geometric fit are accepted for further steps. The candidate fragments are then filtered according to the loop sequence, selecting those with a positive BLOSUM62 score. Candidates that clash with rest of the structures are eliminated. We then select 5 candidates based on their local conformation profile (see [2]) and 5 candidates based on their flank RMSD.
Given the 10 candidates, 3D models of the loop candidates are built by superimposing the candidate linkers on the flanks, and repositioning the side chains using oscar-star [5]. Local geometry refinement is performed by minimizing the conformation of the entire model using Gromacs [6].
In loop remodeling mode, the DaReUS-Loop web server achieves an average top 10 accuracy of 2.00 Å / 2.35 Å on the CASP11 (40 cases) / CASP12 (46 cases) benchmarks. These numbers are within 0.1 Å of the published DaReUS-Loop protocol, and compare favorably to the initial homology model (2.94 Å / 3.52 Å), the Galaxy-PS2 loop modeling server (2.34 Å / 2.88 Å, top 10) and the Rosetta NGK protocol (2.59 Å / 2.99 Å, top 10) on the same benchmarks. We also compared the three modes and observed that the results in modeling (2.49 Å / 2.57 Å) or advanced modeling (2.09 Å / 2.26 Å) modes are somewhat less accurate than remodeling (2.10 Å / 2.18 Å) mode on the CASP11 (48 cases) / CASP12 (50 cases) benchmarks.
DaReUS-Loop has been tested successfully using various OS / browser combinations, including:
[1] Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat, Weissig H, Shindyalov IN, Bourne PE.
The Protein Data Bank
Nucleic Acids Research, 2000; 28: 235-242.
[2]Karami Y, Guyon F, De Vries S, Tufféry P.
DaReUS-Loop: accurate loop modeling using fragments from remote or unrelated proteins.
Sci Rep. 2018; Sep 12;8(1):13673. doi: 10.1038/s41598-018-32079-w.
[3] Guyon F, Tufféry P.
Fast protein fragment similarity scoring using a Binet-Cauchy kernel.
Bioinformatics. 2014 Mar 15;30(6):784-91. doi: 10.1093/bioinformatics/btt618. Epub 2013 Oct 27.
[4] Guyon F, Martz F, Vavrusa M, Bécot J, Rey J, Tufféry P.
BCSearch: fast structural fragment mining over large collections of protein structures.
Nucleic Acids Res. 2015 Jul 1;43(W1):W378-82.
[5] Liang S, Zheng D, Zhang C, Standley DM.
Fast and accurate prediction of protein side-chain conformations.
Bioinformatics. 2011 Oct 15;27(20):2913-4.
[6] Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ.
GROMACS: fast, flexible, and free.
J Comput Chem. 2005 Dec;26(16):1701-18.
[7] Schwede T, Kopp J, Guex N, Peitsch MC
SWISS-MODEL: an automated protein homology-modeling server.
Nucleic Acids Res. 2003; 31 (13): 3381–3385.
[8] Pieper U, Webb BM, Barkan DT, Schneidman-Duhovny D, Schlessinger A, Braberg H, Yang Z, Meng EC, Pettersen EF, Huang CC, Datta RS, Sampathkumar P, Madhusudhan MS, Sjölander K, Ferrin TE, Burley SK, Sali A.
ModBase, a database of annotated comparative protein structure models, and associated resources.
Nucleic Acids Res. 2011 Jan; 39: D465-74.
[9] Söding, J
Protein homology detection by hmm–hmm comparison
Bioinformatics 21, 951–960 (2004)
[10] Zhang, Y & Skolnick, J
Tm-align: a protein structure alignment algorithm based on the tm-score.
Nucleic acids research 33, 2302–2309 (2005)
[11] Eswar, N, Webb, B, Marti‐Renom, MA, Madhusudhan, MS, Eramian, D, Shen, MY, Pieper, U and Sali, A
Comparative protein structure modeling using Modeller.
Current protocols in bioinformatics, 15(1), pp.5-6 (2006)